REVIEW ARTICLE OPEN ACCESS
Epithelial-Mesenchymal Transition in Cancer
Yu Tian1, Noushin Nabavi2, Milad Ashrafizadeh3#
Received 2025 Aug 10
Accepted 2025 Oct 14
Epub ahead of print: December 2025
Published in issue 2026 Feb 15
Correspondence: Milad Ashrafizadeh - Email: dvm.milad1994@gmail.com
The author’s information is available at the end of the article
© 2026 The Author(s). Published by GCINC Press. Open Access licensed under a Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author(s) and source are credited. To view a copy: https://creativecommons.org/licenses/by/4.0/
Abstract
Epithelial-mesenchymal transition (EMT) is a reversible process that enables carcinoma cells to become invasive and therapy-resistant, thereby affecting clinical outcomes such as relapse and treatment failure. In tumors, EMT is triggered by pathways such as TGF-β, Wnt/β-catenin, Notch, PI3K/Akt/mTOR, MAPK, hypoxia, and inflammatory cytokines. These activate EMT-inducing transcription factors, including Snail, Slug, Twist, and ZEB1/2. Noncoding RNAs, like the ZEB-miR-200 axis, also play roles. These changes create intermediate epithelial-mesenchymal states linked to collective migration, stemness, tumor recurrence, and therapy resistance. EMT also promotes immune evasion. Myeloid and stromal cells, especially tumor-associated macrophages and MDSCs, promote EMT and suppress antitumor immunity. EMT reduces antigen presentation, increases immune checkpoints such as PD-L1, and alters chemokines to attract immunosuppressive T cells, helping tumors evade detection. EMT contributes to multidrug resistance by altering cell adhesion and motility and by activating kinases such as STAT3, AXL, and EGFR/ERK. Targeting or reversing EMT can increase tumor sensitivity to treatment and improve outcomes. Combinations of EMT inhibitors (e.g., TGF-β and PI3K inhibitors), epigenetic therapies, and RNA-based reprogramming are being evaluated. New multi-omics and liquid biopsy technologies enable real-time monitoring of EMT status to support more personalized care. Recognizing EMT as a key driver of tumor progression creates new opportunities for targeted treatment.
Keywords: Epithelial-mesenchymal transition (EMT), Cancer metastasis, Drug resistance, Signaling pathways, Tumor microenvironment, Immune evasion, Cancer stemness, Immune checkpoints
1. Introduction
In 2025, the United States is projected to record over 2 million new cancer diagnoses and more than 618,000 cancer-related deaths. Although overall cancer mortality has progressively declined, resulting in an estimated 4.5 million fewer deaths since 1991 due to reduced smoking, earlier detection, and advancements in treatment, significant disparities persist. Notably, Native American and Black populations continue to experience higher mortality rates across several cancer types compared to White populations. Concurrently, while cancer incidence is decreasing among men, it is increasing among women, especially in younger and middle-aged groups. For example, the incidence among women under 50 is now 82% higher than among men, and lung cancer incidence among women under 65 has surpassed that of men. Together, these trends underscore the need for a greater emphasis on prevention, early detection, and equitable healthcare, particularly among underserved populations (1).
The population of cancer survivors in the United States is growing, fueled by an aging population, overall population increase, and advances in early detection and treatment. As of January 1, 2025, there were 18.6 million survivors, with projections exceeding 22 million by 2035. For men, the most common cancers are prostate, melanoma, and colorectal, while for women, breast, uterine, and thyroid cancers are most frequent. More than half of survivors were diagnosed within the past decade, and over 80% are age 60 or older. Despite these advances, significant racial disparities persist. For example, Black patients with early-stage lung or rectal cancer are less likely to undergo surgery compared with White patients. Addressing these inequities with comprehensive, multi-level strategies is therefore essential to ensure quality treatment and survivorship support for all (2).
Metastasis remains a significant challenge in oncology, as it involves a small subset of tumor cells detaching from the primary site, surviving in circulation, and colonizing distant organs to form secondary tumors. Crucially, metastasis is responsible for over 90% of cancer-related deaths, with its invasive and therapy-resistant nature making treatment particularly difficult. Notably, immunotherapy has improved outcomes for some cancers, such as melanoma and lung cancer, yet most metastatic tumors continue to carry poor prognoses. Therefore, understanding the cellular and molecular mechanisms that drive metastasis is essential. One key process is epithelial-mesenchymal transition (EMT), which enables epithelial tumor cells to acquire mesenchymal-like properties. This transition increases their motility, invasiveness, and resistance to therapy, changes that depend on complex networks of signaling pathways, transcription factors, epigenetic regulators, and noncoding RNAs. Furthermore, EMT contributes to immune evasion and chemoresistance (Figure 1).
Given the profound significance of epithelial-mesenchymal transition (EMT) in driving cancer progression, this review comprehensively synthesizes current knowledge regarding the roles of EMT in cancer. Emphasis is placed on its diverse regulatory mechanisms, metastasis, and therapy resistance across multiple tumor types. In addition, the review explores emerging and established strategies to target EMT-related processes and discusses how such interventions may improve clinical outcomes by reducing recurrence and enhancing treatment sensitivity. By combining insights from molecular biology, immunology, and oncology, this review highlights EMT as both a cellular program and a key driver within the tumor microenvironment. Elucidating the complex processes underlying EMT is expected to provide novel perspectives on tumor progression and to support the development of more effective, personalized, and durable therapeutic interventions that can reshape future cancer care.
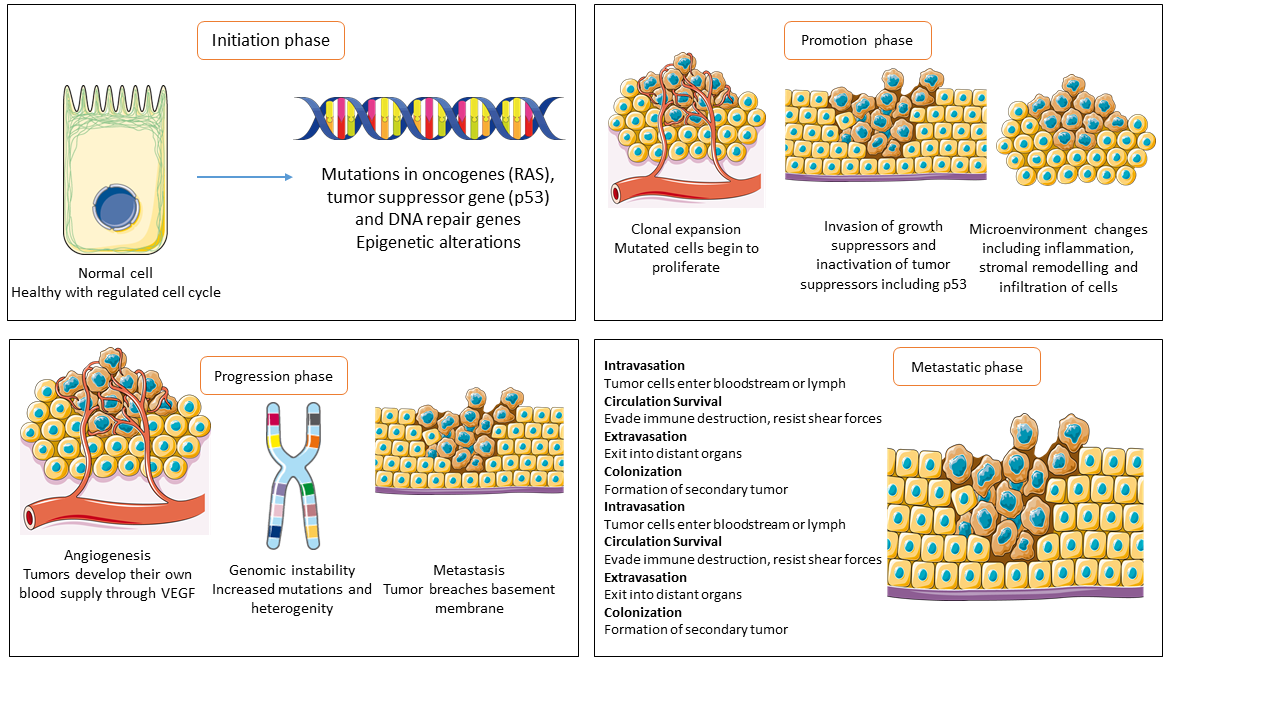
Figure 1: Cancer develops through several phases, each shaped by molecular and cellular events. The initiation phase involves epigenetic changes and oncogene mutations, leading to neoplastic transformation. During promotion, altered cells expand, evade growth suppression, and interact with the microenvironment to aid tumor development. Tumors gain angiogenesis, invasiveness, and genomic instability as they progress. Metastatic cancer cells spread to distant organs, causing most cancer deaths. Understanding molecular circuits, particularly in metastasis, is essential for preventing spread and improving outcomes.
2. EMT mechanism: basics and principles
EMT involves molecular signals within the tumor microenvironment that affect tumor growth, invasion, and metastasis. Enzymes such as MMPs, regulated by TGF-β/Smad signaling, and developmental EMT programs are key to processes such as intravasation, extravasation, and colonization. EMTs are classified into three types: type I (embryogenesis), type II (wound healing/tissue repair), and type III (tumor progression/spread). These subtypes are influenced by the tumor microenvironment, stromal-epithelial interactions, and immune responses, all of which affect therapy response. Stromal fibroblast activation disrupts epithelia and fosters invasion. In development, EMT shapes germ layers. In cancer, similar processes raise vimentin/N-cadherin levels and lower E-cadherin levels, markers of aggressiveness. Wnt and TGF-β/Nodal/Vg1 pathways regulate EMT transcription factors, including Snail, EOMES, and Mesp1/2. Snail reduces E-cadherin and integrins, thereby enabling progression; some effects are reversible. Changes in EMT-related miRNAs, including decreased miR-200 levels, enhance tumor plasticity and metastasis, thereby affecting prognosis. Inhibiting TGF-β (e.g., SB-431542) can block activin- and miR-200-mediated changes, suggesting these pathways as potential therapeutic targets (6).
In normal tissues, EMT is controlled by networks that regulate EMT transcription factors (EMT-TFs) at both transcriptional and post-translational levels (7). Controls include alternative splicing, non-coding RNAs, epigenetic changes, and protein stability (8-14). EMT-TFs activate mesenchymal genes and affect other cellular processes by regulating gene expression (11, 15, 16). EMT promotes tumor growth and spread, and affects tumor initiation and therapeutic responses (16,17). In cancer, EMT is dynamic and reversible, marked by shifting cellular states that mimic embryonic development. EMT and its reverse, mesenchymal-to-epithelial transition (MET), cycle in early development and morphogenesis (7,15,16,18).
EMT refers to the process by which stationary epithelial cells transition into a motile mesenchymal phenotype (19). This phenomenon was first identified in early embryonic development (20), where it plays a crucial role in processes such as gastrulation, neural crest formation, and cardiac development (15, 16). EMT also contributes to essential physiological processes, including wound healing (21) and tissue homeostasis (22). Importantly, abnormal reactivation of EMT is a key driver in the development of pathological conditions, including organ fibrosis and the progression of cancer to metastasis (19). All cells in the human body originate from a single progenitor, with distinct phenotypes arising through the selective activation of specific transcriptomes that drive functional specialization. During embryonic development, epithelial cells exhibit remarkable plasticity, allowing them to switch between epithelial and mesenchymal states via the epithelial-mesenchymal transition (EMT) and its reverse, the mesenchymal-epithelial transition (MET) (23). After organogenesis is complete, epithelial cells adopt specialized functions (24,25).
For many years, it was believed that epithelial cells must undergo terminal differentiation to perform their specialized functions and that this state was irreversible. This view has been overturned by evidence showing that even terminally differentiated epithelial cells can undergo phenotypic changes in response to repair-associated or pathological stress. One of the core mechanisms driving cellular diversity during both development and adulthood is EMT. As highlighted by Robert Weinberg and colleagues (26), EMT plays central roles across various biological contexts, promoting cell dispersion during embryogenesis, driving the generation of mesenchymal cells or fibroblasts in response to tissue injury, and enhancing the invasive and metastatic potential of epithelial cancer cells. Beyond its developmental functions, EMT is also linked to evolutionary processes that enabled the emergence of complex multicellular organisms. In adult tissues, EMT is reactivated during wound healing, tissue regeneration, and the progression of fibrotic diseases, thereby facilitating the generation of mesenchymal cells critical for tissue remodeling. In cancer, genetic alterations in epithelial cells create selective pressures that support both local invasion and systemic spread, processes greatly amplified by EMT, which endows tumor cells with migratory and invasive capacities (23).
EMT is regulated by several key signaling pathways, most notably TGF-β, Notch, and Wnt (27). The tumor microenvironment (TME) further shapes these pathways through external influences, including hypoxia and alterations in microRNA (miRNA) expression (28,29). Despite their differences, these signaling pathways converge on a common set of transcription factors, Snail (SNAI), Zeb, and Twist, that are well known for driving EMT when aberrantly expressed in cancers. Through both canonical and non-canonical routes, TGF-β signaling regulates the expression of transcription factors, including Snail, Zeb, Twist, and Six1 (29). Similarly, Notch signaling activates NF-κB, which in turn promotes the transcription of SNAI1, SNAI2 (also known as Slug), Twist, and Zeb1/2, while also enhancing cytokine secretion and supporting cell survival. The Wnt pathway, frequently dysregulated in cancers, upregulates SNAI1 expression and represses E-cadherin by activating β-catenin. Additionally, environmental stresses, such as hypoxia, disrupt mitochondrial function, triggering HIF-1 activation and elevating Zeb1 expression (27). Figure 2 provides an overview of the EMT mechanism and its associated factors. In this process, epithelial cells transition into mesenchymal cells, characterized by the upregulation of N-cadherin, vimentin, and fibronectin, along with increased cell motility, accompanied by the downregulation of E-cadherin. EMT-inducing transcription factors, including SNAIL, SLUG, TWIST1/2, and ZEB1/2, play a central role in driving this transition. Both positive and negative regulators, such as TGF-β, Wnt/β-catenin, miRNAs, Hedgehog, Notch, and hypoxia-inducible factor (HIF), further modulate the process. Additionally, feedback loops, such as the interaction between ZEB1 and the miR-200 family, contribute to the fine regulation of EMT.
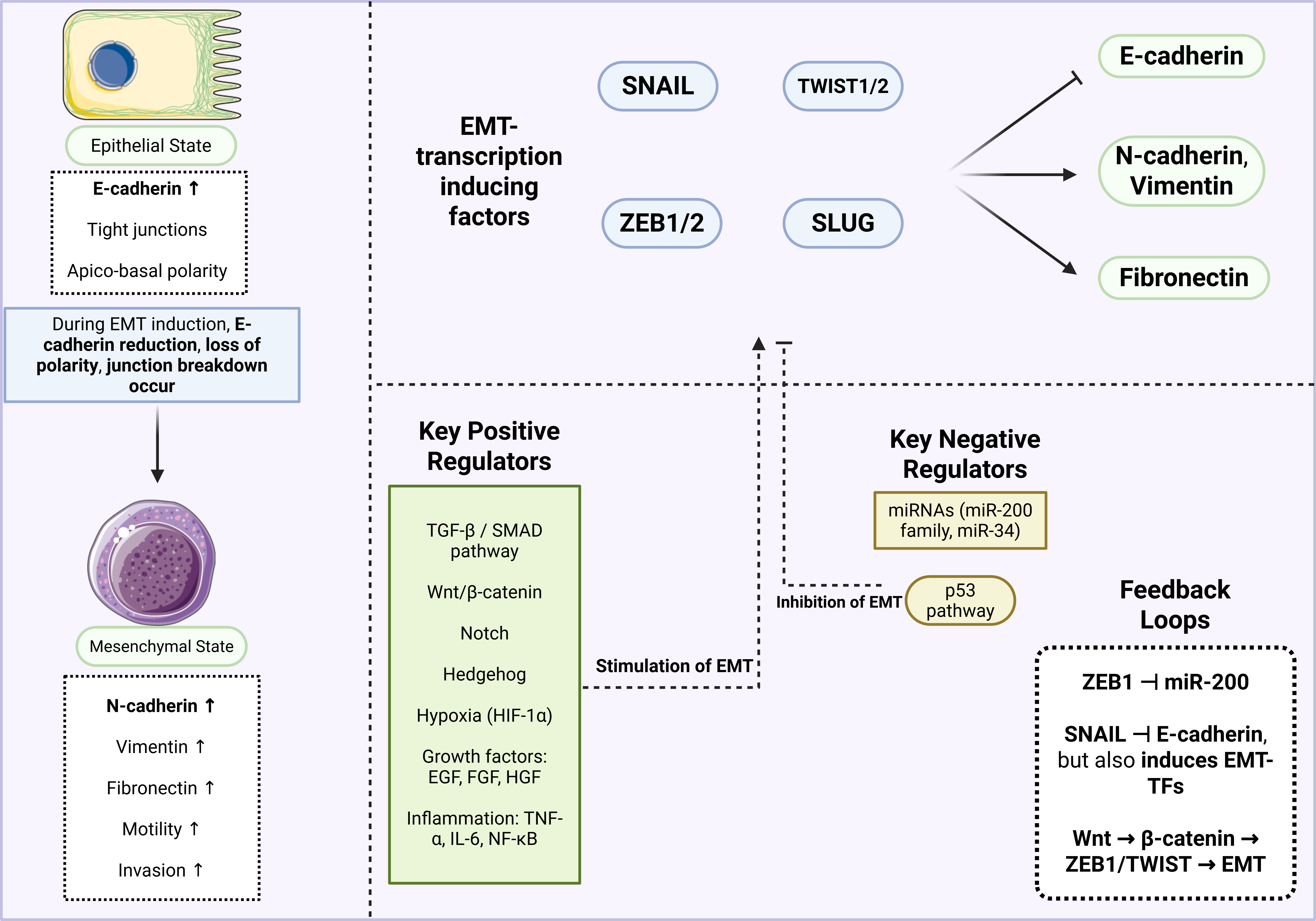
Figure 2: An overview of the EMT mechanism. In the epithelial state, cells exhibit high E-cadherin expression, tight junctions, and apico-basal polarity. During EMT, however, cells transition to a mesenchymal state characterized by upregulation of N-cadherin, vimentin, and fibronectin, thereby increasing motility and invasiveness. Key transcription factors, including SNAIL, SLUG, TWIST, and ZEB, drive this transition, while both positive and negative regulators modulate the process. Given EMT’s critical role in cancer progression, inhibiting it offers promising opportunities for novel therapeutic strategies.
During EMT, cells activate multiple interconnected signaling pathways that form a complex regulatory network of genes, growth factors, and cytokines. Although most illustrations capture only part of this crosstalk, it highlights the intricate control of EMT. A hallmark of EMT is the active reorganization of the cytoskeleton, a process critical for enabling cell migration and motility during metastasis. TGF-β is a key initiator of EMT, acting through both canonical and non-canonical signaling pathways. In the canonical pathway, TGF-β activates SMAD protein complexes that control transcription factors involved in apoptosis and cell adhesion. Through the non-canonical pathway, TGF-β engages the PI3K/Akt signaling cascade, promoting protein synthesis, cell proliferation, and resistance to anoikis. It also activates Rho GTPases and JNK signaling, promoting junction disassembly and cytoskeletal reorganization, key steps in invasion and motility. The MAPK/ERK pathway may be triggered either by TGF-β signaling or through integrin-mediated cell-cell interactions, leading to PI3K activation and the enhancement of mesenchymal characteristics. Similarly, receptor tyrosine kinases (RTKs) activated by growth factors stimulate MAPK/ERK signaling, thereby enhancing mesenchymal characteristics. Additional pathways, including Wnt, Hippo, and Notch, further contribute to EMT by downregulating adhesion molecules, promoting cell detachment, and inhibiting anoikis, thereby amplifying the migratory and invasive potential of cells undergoing EMT (30).
In metastatic cells undergoing EMT, transcription factors such as Snail, Twist, and Zeb are upregulated (31). Snail is strongly regulated by TGF-β signaling, which downregulates cadherin-16 and HNF-1β, key components of the epithelial phenotype, thereby driving EMT (32). Cancer cells must overcome the pro-apoptotic effects of TGF-β to survive, and EMT-associated transcription factors play a central role in this adaptation. Snail, for example, enhances cancer cell survival by upregulating anti-apoptotic proteins such as Akt and Bcl-xL, effectively protecting cells from TGF-β-induced apoptosis (33). Interestingly, while promoting survival, Snail also suppresses cell cycle progression by downregulating Cyclin D2, consistent with the reduced proliferation observed during EMT-associated differentiation (34).
Additionally, the insulin-like growth factor receptor (IGF-1R) contributes to EMT induction. In mammary epithelial cells, IGFR activates NF-κB and Snail, facilitating the transition to a mesenchymal phenotype. In prostate cancer, IGFR signaling has been linked to increased Zeb expression and activation of latent TGF-β1, further reinforcing EMT programs and promoting tumor progression (31,35,36).
3. EMT in cancer drug resistance
Chemoresistance is a significant challenge in the treatment of many cancers, with increasing evidence underscoring the critical roles of the TME, EMT, and key signaling pathways in driving drug resistance and tumor progression. In pancreatic cancer (PC), pancreatic stellate cells (PSCs) secrete hepatocyte growth factor (HGF), which activates the c-Met/PI3K/Akt pathway in PC cells (PCCs), promoting EMT and inhibiting apoptosis, thereby contributing to gemcitabine resistance (37). In PCa bone metastases, bone-derived TGF-β induces the acetylation of KLF5, enhancing resistance to docetaxel and promoting metastasis through CXCR4-mediated IL-11 and SHH/IL-6 paracrine signaling. This process can be therapeutically reversed by targeting CXCR4 (38). In ovarian cancer, TGFβ1 enhances cancer stem cell (CSC)-like properties and drives EMT through both Smad-dependent and Smad-independent pathways, with ZEB1 as a key transcription factor, ultimately leading to increased resistance and tumor heterogeneity (39). In bladder cancer, CircPTK2 interacts with PABPC1 to stabilize SETDB1 mRNA, thereby promoting EMT, enhancing metastasis, and driving gemcitabine resistance through the circPTK2/PABPC1/SETDB1 axis (40). In gastric cancer, the ginsenoside derivative Compound K overcomes oxaliplatin resistance by suppressing the PI3K/Akt pathway, reversing EMT, and decreasing drug efflux through downregulation of P-gp, thereby demonstrating potent antitumor activity in vitro and in vivo (41). In gastric cancer, the ginsenoside derivative Compound K reduces oxaliplatin resistance by inhibiting the PI3K/Akt pathway, reversing EMT, and lowering drug efflux via downregulation of P-gp, demonstrating strong anti-tumor effects in both in vitro and in vivo models. The unfavorable prognosis of pancreatic ductal adenocarcinoma (PDAC) is partly attributed to its resistance to chemotherapy. Glutathione peroxidase-1 (GPx1), a key antioxidant enzyme implicated in several cancers, remains unclear in PDAC. Tissue microarray analyses revealed that higher GPx1 expression was associated with shorter overall survival in patients with PDAC. GPx1 inhibition promoted a mesenchymal phenotype and enhanced gemcitabine resistance in both in vitro and in vivo models. RNA sequencing demonstrated that this effect was mediated, in part, through the activation of reactive oxygen species (ROS)-induced Akt/glycogen synthase kinase 3β (GSK3β)/Snail signaling. Furthermore, low GPx1 expression was associated with worse outcomes in PDAC patients treated with GEM-based adjuvant chemotherapy, which was not observed in those receiving fluoropyrimidine-based regimens (42).
Prostate cancer is an aggressive malignancy characterized by extensive extracellular matrix (ECM) deposition and a pronounced EMT phenotype, both of which contribute to chemoresistance in desmoplastic tumors. To overcome these barriers, an oncolytic adenovirus co-expressing decorin and a soluble Wnt decoy receptor (HEmT-DCN/sLRP6) was evaluated in an orthotopic pancreatic xenograft model using Mia PaCa-2 cells implanted in athymic nude mice. Systemic administration of HEmT-DCN/sLRP6 elicited robust anticancer and antimetastatic effects. Histological analyses revealed marked ECM degradation, enhanced apoptosis, increased viral dissemination within the tumor, and effective inhibition of Wnt/β-catenin signaling. Treatment with HEmT-DCN/sLRP6 suppressed EMT, reduced tumor cell proliferation, and significantly inhibited PC metastasis. Additionally, HEmT-DCN/sLRP6 increased gemcitabine sensitivity in pancreatic tumor xenografts and patient-derived tumor spheroids by facilitating greater drug penetration and distribution (43).
Chemoresistance remains a significant obstacle in cancer therapy, frequently driven by EMT, dysregulated signaling pathways, and CSC properties across multiple tumor types. In gastric cancer, the transcription factor GLI2, an essential mediator of the Hedgehog pathway, is upregulated in EMT-type GC and contributes to cisplatin resistance by promoting EMT through a novel GLI2/DEC1/ZEB1 axis (44). In cervical cancer, iASPP drives EMT and cisplatin resistance by upregulating miR-20a in a p53-dependent manner. This, in turn, suppresses the tumor suppressors FBXL5 and BTG3, resulting in increased cell invasion and a poor prognosis (45).In PC, EMT drives gemcitabine resistance through multiple mechanisms: cadherin switching from E-cadherin to N-cadherin reduces ENT1 expression and drug uptake, whereas EpCAM counteracts EMT and restores chemosensitivity (46).In gemcitabine-resistant PCCs, EMT and CSC phenotypes are sustained by glycolysis, which maintains low ROS levels and promotes DCLK1 production; inhibiting glycolysis or increasing ROS disrupts this resistance (47). Another study highlights the role of LAMC2, which is linked to poor prognosis in PDAC and promotes gemcitabine resistance by driving EMT and regulating ATP-binding cassette (ABC) transporters (48).
These results underscore the key role of EMT-related pathways, such as GLI2, iASPP/miR-20a, cadherin switching, DCLK1, and LAMC2, in promoting chemoresistance, making them promising targets for therapies to overcome drug resistance in gastrointestinal and gynecological cancers. Galectin-1 (Gal-1) also emerges as a key mediator of tumor progression and chemoresistance; however, its roles in EMT and sorafenib resistance in hepatocellular carcinoma (HCC) remain uncertain. Recent evidence shows that elevated Gal-1 expression accelerates HCC development and reduces sorafenib sensitivity through αvβ3-integrin-mediated activation of the AKT pathway. Additionally, Gal-1 promotes EMT in HCC cells through the PI3K/AKT signaling pathway. Clinically, high Gal-1 levels are associated with poorer survival and reduced sorafenib efficacy in patients with HCC, suggesting that Gal-1 may be a therapeutic target and a prognostic biomarker for predicting sorafenib response in HCC (49).
EMT has emerged as a key mechanism driving chemoresistance in multiple cancers, including head and neck squamous cell carcinoma (HNSCC), colorectal cancer (CRC), and non-muscle invasive bladder cancer (NMIBC). In HNSCC, resistance to the EGFR inhibitor gefitinib is associated with EMT, exemplified by the UMSCC81B cell line variant (81B-Fb), which exhibits fibroblast-like morphology, reduced E-cadherin expression, and elevated vimentin and Snail levels (50). The EMT phenotype is driven by EGFR downregulation, facilitated by enhanced ubiquitination and activation of the Akt/GSK-3β/Snail signaling pathway. This effect can be partially reversed by the PI3K inhibitor LY294002 or by EGFR overexpression, indicating a pathway-specific mechanism underlying gefitinib resistance. In the context of MEK1/2 inhibitor (MEKi) resistance, amplification of oncogenic BRAFV600E or KRASG13D restores ERK1/2 signaling; however, only BRAFV600E-mediated resistance is reversible upon drug withdrawal (51). Reversibility depends on p57KIP2 activation, which induces G1 arrest and senescence, ultimately leading to loss of BRAFV600E amplification and renewed sensitivity to MEKi (Figure 3).
In contrast, KRASG13D amplification sustains ERK1/2 activation, driving a ZEB1-dependent EMT and persistent chemoresistance, rendering drug therapies ineffective. In CRC, high expression of the EMT-inducing transcription factor ZEB2 is associated with a poor prognosis and resistance to adjuvant FOLFOX therapy. ZEB2 fosters chemoresistance by upregulating the nucleotide excision repair pathway, particularly through ERCC1, thereby enhancing tumor survival against oxaliplatin. Consistently, tumors overexpressing ERCC1 demonstrated oxaliplatin resistance in vivo, highlighting the ZEB2-ERCC1 axis as a critical contributor to treatment failure and disease recurrence (52). In NMIBC, CXCL5 overexpression has been shown to drive mitomycin C resistance by inducing EMT and activating the NF-κB pathway (53). Exogenous CXCL5 reduced drug sensitivity in parental cells, whereas its knockdown restored responsiveness and suppressed EMT/NF-κB signaling. Collectively, these observations highlight the crucial role of EMT and its regulatory networks in driving chemoresistance across multiple cancer types by modulating growth factor receptors, transcriptional reprogramming, and cytokine signaling, underscoring the importance of targeting EMT-related pathways to overcome therapeutic resistance (Figure 3).
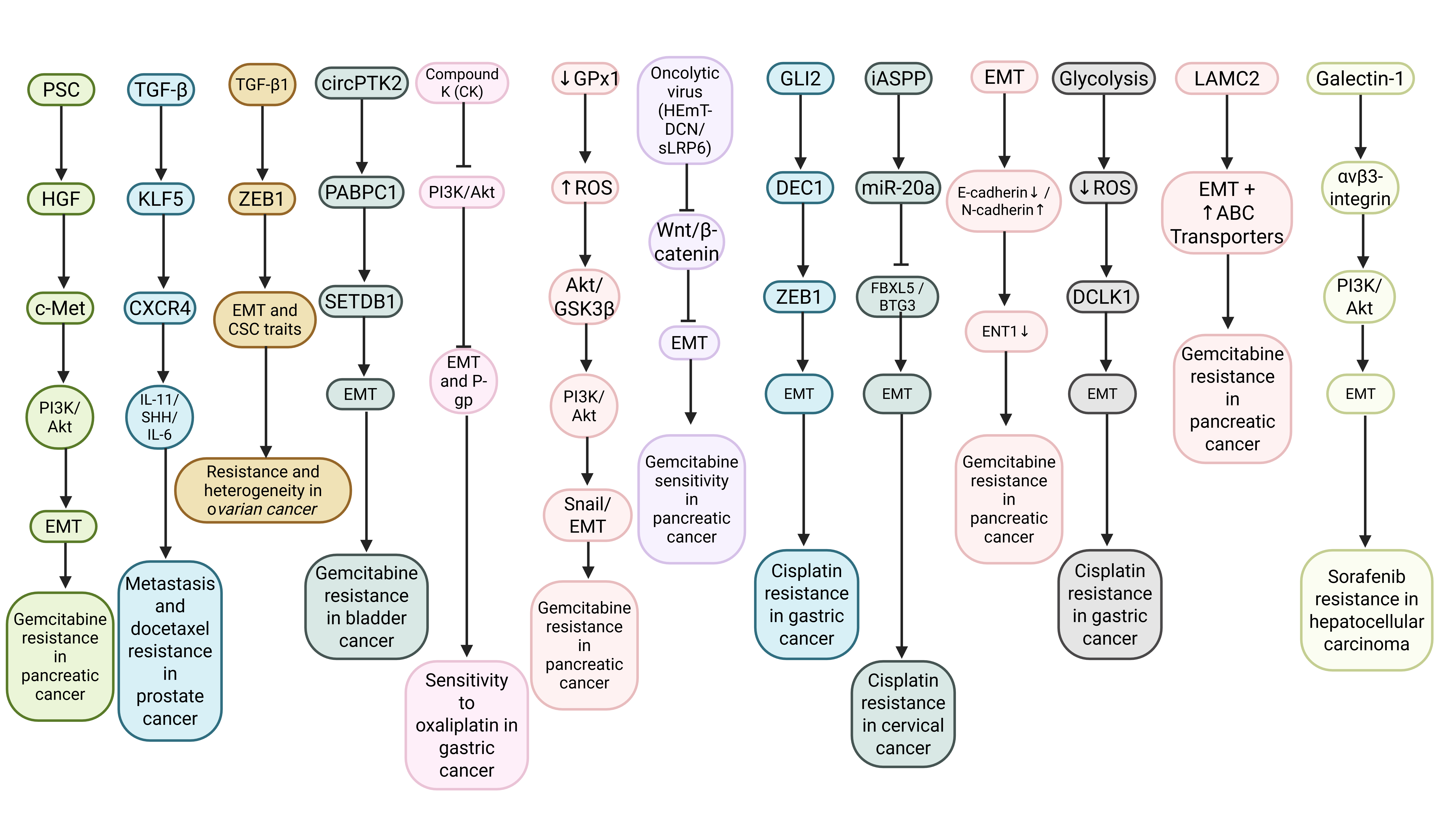
Figure 3: The EMT mechanism in cancer drug resistance. The figure illustrates key interactions among TME components, oncogenic signaling pathways (PI3K/Akt, Wnt/β-catenin), transcription factors (ZEB1, Snail, DEC1), metabolic reprogramming, and drug transport modulation. Upstream stimuli, including growth factors, cytokines, non-coding RNAs, and ECM remodeling, converge on EMT and CSC-like phenotypes, ultimately promoting reduced drug uptake, enhanced survival signaling, and increased metastatic potential. Inhibitory arrows depict therapeutic interventions targeting these pathways to restore chemosensitivity.
Table 1 describes how the acquisition of chemoresistance in cancer is closely linked to EMT induction, characterized by the loss of epithelial markers, upregulation of mesenchymal markers, and activation of EMT-associated transcription factors, including Snail, Slug, Twist, and the ZEB family. This transition frequently engages signaling pathways including STAT3, PI3K/Akt/mTOR, MAPK, ATM/JAK/STAT, and EGFR-driven cascades, which collectively enhance migration, invasion, stemness, and survival under therapeutic stress. EMT regulators promote drug resistance by reprogramming tumor cells into mesenchymal phenotypes, thereby diminishing their sensitivity to chemotherapeutic agents and increasing their metastatic potential. Conversely, inhibition or silencing of EMT drivers or their upstream regulators has been shown to reverse the mesenchymal state, restore epithelial traits, and re-sensitize cancer cells to therapy. These findings highlight EMT-associated signaling networks as central mediators of therapeutic resistance, underscoring their value as targets for improving treatment efficacy and clinical outcomes.
Table 1: Chemoresistance in cancer and relation to EMT induction.
| Cancer Type | Drug/Resist Context | EMT Markers/Features | Key EMT Regulator | Signaling Pathway | Functional Outcome | Ref |
|---|---|---|---|---|---|---|
| Gastric Cancer | Cisplatin resistance: eIF5A2 overexpression reduced sensitivity | ↓ E-cadherin, ↓ β-catenin, ↑ N-cadherin, ↑ Vimentin; EMT induction | Twist, eIF5A2 | EMT | eIF5A2 promotes EMT → decreased sensitivity to cisplatin; knockdown of eIF5A2 or Twist restores sensitivity | (56) |
| Ovarian Cancer | Cisplatin resistance in A2780cis cells | ↓ E-cadherin, ↑ vimentin, ↑ Snail, ↑ Slug; spindle morphology, pseudopodia formation, increased migration and invasion |
Snail, Slug, Twist2, ZEB2, ZEB1, Twist1 |
EMT | EMT promotes cisplatin resistance, invasion, and migration; knockdown of Snail/Slug reverses EMT and re-sensitizes cells to cisplatin | (57) |
| Lung Cancer | Acquired resistance to cisplatin in A549 cells | ↑ N-cadherin, ↓ E-cadherin in WT-CisR; STAT3 KO reverses EMT to epithelial phenotype | STAT3 |
STAT3 mTOR mTORC Akt) |
STAT3 promotes EMT and cisplatin resistance via mTOR activation; STAT3 knockout prevents resistance, reverses EMT, and sensitizes cells to cisplatin and rapamycin | (58) |
|
Stomach Adeno carcinoma |
Cisplatin resistance associated with high Rab31 expression | ↓ E-cadherin, ↑ Vimentin, ↑ MMP-2, ↑ MMP-9; increased migration, colony formation, and apoptosis resistance | Twist1, STAT3, MUC-1 | Rab31 → STAT3 → MUC-1 ↓ → Twist1 ↑ → EMT | Rab31 promotes EMT and cisplatin resistance via Twist1; knockdown restores drug sensitivity and reduces metastasis | (59) |
| NSCLC | Cisplatin resistance in A549 and H157 cells; upregulated ATM expression | ↓ E-cadherin, ↑ N-cadherin, ↑ Vimentin, ↑ Snail, ↑ Zeb1, ↑ Twist, ↑ VEGF; mesenchymal morphology; increased invasion & migration |
ATM, PD-L1, JAK1/2, STAT3 |
ATM → JAK1/2 → STAT3 → PD-L1 → EMT | ATM promotes EMT and metastasis, leading to cisplatin resistance; inhibition of ATM reverses EMT and reduces metastasis both in vitro and in vivo | (60) |
| TNBC | Cisplatin (CIS) resistance; tested with sulforaphane (SFN) combination therapy | ↑ E-cadherin, Claudin, ZO1; ↓ N-cadherin, Vimentin, β-catenin, MMP-2/9; ↓ EMT-TFs: Snail, Slug, ZEB1; shift from mesenchymal to epithelial |
Snail, Slug, ZEB1, SIRT1, SIRT7 |
TGF-β1 → SIRTs (SIRT1/2/3/5/7) → EMT; SFN + CIS inhibit SIRTs and reverse histone deacetylation at E-cadherin promoter | SFN + CIS synergistically inhibit growth, trigger S-phase arrest, curb migration, invasion, reverse EMT, suppress sirtuins, restore E-cadherin, and overcome cisplatin resistance/metastasis. | (61) |
| Ovarian Cancer | Cisplatin resistance mediated by STAT3 phosphorylation at Tyr705 | ↓ E-cadherin; ↑ Vimentin, ↑ Slug, ↑ Snail; enhanced migration, invasion, and mesenchymal morphology |
STAT3 (pY705), Slug, Snail |
STAT3 → PI3K/AKT/mTOR & MAPK → inhibition of ER stress-induced autophagy | STAT3 activation drives EMT, proliferation, migration, invasion, and cisplatin resistance; STAT3 inhibition or p53 activation reverses EMT and restores sensitivity. RAS cooperates with STAT3 in resistance and tumorigenesis. | (62) |
| HCC | Doxorubicin resistance: higher ARK5 expression correlates with lower drug sensitivity | ↓ E-cadherin; ↑ Vimentin under doxorubicin or hypoxia; reversed upon ARK5 knockdown | ARK5, TWIST | ARK5 → EMT (via TWIST); EMT likely mediates drug resistance | ARK5 knockdown increases doxorubicin sensitivity under normoxia/hypoxia and reverses EMT. TWIST knockdown blocks EMT and prevents ARK5-driven resistance. ARK5 induces resistance by promoting EMT, but not when EMT is already blocked. | (63) |
| TNBC | Doxorubicin resistance in MDA-MB-231cells through chronic exposure | ↓ E-cadherin; ↑ N-cadherin, β-catenin, ICAM-1; increased CD44 and OCT3/4; mesenchymal morphology | ↑ EGFR | EGFR → AKT/ERK signaling; EGFR downstream pathways promote EMT, CSC, drug resistance | DRM cells enhance proliferation, migration, invasion, adhesion, CSC expansion, EMT traits, and doxorubicin resistance. This resistance spreads to parental cells via autocrine signals, while EGFR‑AKT/ERK pathways maintain EMT and CSC features, driving resistance. | (64) |
4. Major molecular pathways regulating EMT in cancer
4.1 PI3K/Akt/mTOR and PTEN signaling pathway
EMT, a key mechanism in tumor invasion and metastasis, provides valuable insights for the development of novel cancer therapies. Sotetsuflavone, a natural compound derived from Cycas revoluta, has shown notable anticancer activity during the early stages of tumor progression. A study investigated the antimetastatic effects of sotetsuflavone in vitro, demonstrating that it suppresses metastasis and EMT in A549 non-small-cell lung cancer (NSCLC) cells. The inhibitory effect was marked by increased E-cadherin expression and decreased levels of N-cadherin, vimentin, and Snail. Mechanistic analysis revealed that HIF-1α plays a central role in mediating sotetsuflavone's anti-metastatic activity. The compound modulated VEGF signaling by downregulating VEGF and upregulating angiostatin, while also reducing MMP-9 and MMP-13 expression. Furthermore, sotetsuflavone inhibited HIF-1α activity by suppressing the PI3K/AKT and TNF-α/NF-κB pathways (63).
EMT and angiogenesis are recognized as key mechanisms in cancer progression. Curcumin, known for its favorable safety profile, has been widely studied in both preclinical and clinical settings for cancer prevention. However, its role in regulating EMT and angiogenesis in lung cancer remains insufficiently understood. In this study, curcumin suppressed induced cell migration and EMT-related morphological changes in A549 and PC-9 lung cancer cells. Pretreatment with curcumin inhibited HGF-induced c-Met phosphorylation and downstream activation of Akt, mTOR, and S6 proteins, effects comparable to those of the c-Met inhibitor SU11274, the PI3K inhibitor LY294002, and the mTOR inhibitor rapamycin. Overexpression of c-Met further confirmed that curcumin blocks EMT in lung cancer cells by interfering with the c-Met/Akt/mTOR signaling pathway. Additionally, curcumin markedly suppressed PI3K/Akt/mTOR activity, induced apoptosis, and inhibited migration and tube formation in human umbilical vein endothelial cells (HUVECs) stimulated by HGF. In a murine experimental model, curcumin reduced HGF-driven tumor growth, accompanied by increased E-cadherin expression and decreased levels of vimentin, CD34, and VEGF (64).
The intricate dynamics of cancer progression consistently highlight EMT as a central driver of tumor dissemination and poor prognosis across diverse malignancies. Among its regulators, the PI3K/AKT signaling pathway has emerged as a critical hub linking oncogenic proteins to cancer aggressiveness. In CRC, FAT4, a well-known tumor suppressor, was found to be downregulated in tumor tissues and shown to inhibit invasion, migration, and proliferation by enhancing autophagy and suppressing EMT through the PI3K/AKT/mTOR and PI3K/AKT/GSK-3β pathways. In bladder cancer, TEAD4 was identified as an oncogene strongly associated with higher tumor grade and poorer survival; it promoted EMT and metastasis in both in vitro and in vivo models by activating the PI3K/AKT pathway, whereas pharmacological inhibition of this pathway reduced TEAD4-driven malignancy. Similarly, transforming acidic coiled-coil protein 3 (TACC3) has been shown to induce EMT and enhance transformation, proliferation, and invasion across multiple cancers by activating both the PI3K/AKT and ERK pathways, suggesting it is a viable therapeutic target. In thyroid cancer, the hematopoietic PBX-interacting protein (HPIP) functions as an oncogene, and its silencing effectively suppresses proliferation, migration, and EMT by inhibiting PI3K/AKT signaling. Together, these findings underscore the diverse yet interconnected roles of FAT4, TEAD4, TACC3, and HPIP in regulating EMT and metastasis via the PI3K/AKT axis, positioning them as promising therapeutic targets to halt tumor progression and improve patient outcomes (65-68).
The Williams syndrome transcription factor (WSTF), encoded by the BAZ1B gene, was initially identified as a hemizygously deleted gene in individuals with Williams syndrome. WSTF functions in transcription, replication, chromatin remodeling, and the DNA damage response, and acts as a tyrosine protein kinase. In lung cancer cell lines A549 and H1299, WSTF overexpression was shown to enhance proliferation, colony formation, migration, and invasion. In vivo studies using mouse xenograft models further confirmed that elevated WSTF expression drives tumor growth and invasiveness. Transcriptomic profiling, validated by qRT-PCR, revealed that WSTF overexpression significantly upregulates EMT markers, including fibronectin (FN1), and EMT-inducing genes, including Fos and CEACAM6. These molecular changes were characterized by reduced E-cadherin and increased N-cadherin and FN1 expression at both mRNA and protein levels, along with morphological features consistent with EMT. Mechanistically, WSTF was found to activate PI3K/Akt and IL-6/STAT3 signaling pathways. Pharmacological inhibition with the PI3K inhibitor ZSTK474 or the STAT3 inhibitor niclosamide reduced WSTF-induced proliferation, migration, and invasion, while lowering the levels of phosphorylated Akt, phosphorylated STAT3, and IL-6. Importantly, these inhibitors also restored epithelial markers and suppressed EMT-related transcription factors, including Snail, Slug, Twist, and CEACAM6, in WSTF-overexpressing A549 cells (69).
The ubiquitin-binding enzyme E2T (UBE2T), a member of the E2 ubiquitin-proteasome family, has been implicated in carcinogenesis across multiple malignancies, but its role in ovarian cancer has only recently been clarified. Emerging evidence shows that UBE2T is markedly overexpressed in ovarian cancer tissues, particularly in cases with BRCA mutations, and this elevated expression strongly correlates with poor patient prognosis. Immunohistochemical analysis confirmed UBE2T upregulation, representing the first report linking its expression to BRCA-mutated ovarian cancer. Functional studies demonstrated that silencing UBE2T significantly reduced ovarian cancer cell proliferation and invasion, while suppressing EMT and downregulating PI3K-AKT signaling. Conversely, treatment with the mTOR activator MHY1485 reactivated PI3K-AKT signaling and largely restored the proliferative and invasive capacity of UBE2T-deficient cells. In vivo tumorigenesis experiments in nude mice further validated that UBE2T knockdown inhibited tumor growth and EMT in tumor tissues. Collectively, these findings suggest that UBE2T acts as an oncogenic driver in ovarian cancer by regulating EMT through the PI3K-AKT pathway, potentially in cooperation with BRCA mutations. Thus, UBE2T holds promise as a novel biomarker for early diagnosis, prognosis, and targeted therapy in ovarian cancer, and its inhibition may represent a viable therapeutic strategy (70).
KIAA1199 is frequently upregulated in various cancers, including NSCLC, where its expression is associated with aggressive tumor behavior and poor patient survival. Analyses of preserved clinical specimens revealed significantly higher KIAA1199 mRNA and protein levels in NSCLC tissues than in adjacent normal tissues, and higher expression was an independent predictor of overall survival. Functional studies in NSCLC cell lines demonstrated that KIAA1199 regulates EMT by altering the expression of EMT markers, EMT-inducing transcription factors, and associated signaling molecules. Gene silencing or overexpression of KIAA1199 correspondingly modulated EMT pathway activity, highlighting its regulatory role in this process. In a mouse xenograft model, KIAA1199 knockdown significantly suppressed tumor growth and enhanced treatment response. Collectively, these findings indicate that KIAA1199 acts as a prognostic biomarker and therapeutic target in NSCLC by modulating EMT signaling (71).
EMT plays a central role in promoting tumor progression across various cancers, including colorectal and lung cancer, and is closely controlled by the tumor suppressor PTEN. In CRC cells, PTEN loss induces EMT through Akt activation and alters chromatin accessibility by phosphorylating EZH2 at Ser21. This modification converts EZH2 from a repressor to an activator, disrupting its association with the PRC2 component SUZ12 and reducing H3K27me3 levels. The resulting epigenetic reprogramming activates transcription factors such as AP1, thereby promoting EMT (72). In colon cancer cells, treatment with 2′-benzoyloxycinnamaldehyde (BCA) suppresses EMT and invasion by upregulating EGR1, which in turn enhances PTEN expression (73). Silencing EGR1 or PTEN diminishes the inhibitory effects of BCA on EMT markers, including Snail and β-catenin. In lung cancer, CRISPR/Cas9-mediated PTEN deletion enhances proliferation, migration, invasion, and metastasis in vivo, accompanied by increased phosphorylation of Akt and GSK-3β, as well as nuclear translocation of β-catenin, Snail, and Slug (74). PTEN-deficient cells also display morphological changes characteristic of EMT, including reduced cell-cell adhesion and increased pseudopodia formation. These findings highlight the crucial role of PTEN in preserving epithelial integrity and inhibiting EMT across various cancers, primarily by regulating the PI3K/Akt signaling pathway and modulating β-catenin localization. EMT, a critical driver of cancer progression and metastasis, is controlled by multiple molecular factors, with phosphatase and tensin homolog (PTEN) acting as a central modulator in many tumor types. In gastric cancer, the oncogenic enhancer of zeste homolog 2 (Ezh2) is overexpressed. It promotes tumor growth, EMT, and CSC-like traits by suppressing PTEN and activating the Akt pathway, with the PTEN/Akt axis mediating these effects (75). In endometrial carcinoma, PTEN overexpression induces EMT and CSC traits by activating nuclear β-catenin and Slug, while concurrently reducing proliferation and promoting apoptosis, particularly in ERα/EBP50-deficient contexts (76). PRL-3, a metastasis-associated phosphatase, promotes EMT by activating the PI3K/Akt pathway and suppressing PTEN expression, thereby reducing epithelial marker expression, reorganizing the actin cytoskeleton, and enhancing cell motility; these effects depend on its phosphatase activity (77). In tongue squamous cell carcinoma (TSCC), PTEN overexpression suppresses proliferation, invasion, and EMT by downregulating Hedgehog signaling, thereby reinforcing its tumor-suppressive role (78). Across these studies, PTEN emerges as a key suppressor of EMT and metastasis, primarily by inhibiting the PI3K/Akt pathway and its downstream transcription factors, such as Snail and Slug. However, in specific contexts, such as endometrial carcinoma, PTEN can paradoxically promote EMT and CSC traits via alternative signaling routes, underscoring its complex, context-dependent role. PTEN and its regulatory network, including modulators such as Ezh2, PRL-3, and EGR1, therefore represent compelling therapeutic targets for blocking EMT and limiting cancer progression. Chronic inflammation is a well-established contributor to cancer progression, with cytokines such as tumor necrosis factor-α (TNF-α) playing a significant role in enhancing tumor invasiveness. In human colon cancer cell lines HCT116 and SW480, siRNA-mediated knockdown of the tumor suppressor PTEN significantly increased cell invasion and motility, as demonstrated in Boyden chamber and scratch assays. When PTEN-deficient cells were exposed to TNF-α, their invasive and migratory capacities were further enhanced, suggesting a synergistic effect between PTEN loss and TNF-α-mediated inflammatory signaling. Mechanistically, PTEN silencing led to nuclear accumulation of β-catenin and upregulation of its downstream targets, c-Myc and cyclin D1, both of which are implicated in tumor development. These findings align with clinical observations linking PTEN deficiency to advanced stages of CRC and suggest that genetic alterations within tumor cells, combined with inflammatory cues from the microenvironment, cooperatively drive enhanced invasion and metastasis (79).
Loss of PTEN or activation of the PI3K/AKT signaling pathway is associated with the progression and metastasis of human prostate cancer (PCa). However, in preclinical mouse models, PTEN deletion alone does not fully reproduce the extensive metastatic burden commonly observed in advanced human disease. Analysis of human prostate cancer (PCa) tissue microarrays revealed substantial activation of the RAS/MAPK pathway in both primary tumors and metastatic lesions. To further investigate, mice carrying a conditionally activatable K-ras (G12D) allele were crossed with a prostate-specific PTEN knockout model. At the same time, RAS activation alone was insufficient to initiate PCa; its combination with PTEN loss markedly accelerated disease progression, driving robust EMT and widespread macrometastasis with complete penetrance. Within these compound mutant prostates, a distinct population of stem/progenitor cells with mesenchymal features was identified, displaying high metastatic potential upon orthotopic transplantation. Importantly, pharmacological inhibition of RAS/MAPK signaling with the MEK inhibitor PD325901 significantly reduced metastasis driven by these stem/progenitor cells. Collectively, these findings demonstrate that RAS/MAPK activation functions as a critical cooperating driver alongside PTEN/PI3K/AKT pathway disruption, suggesting that dual targeting of these pathways may provide an effective strategy for preventing metastatic Prostate Cancer (80).
4.2 STAT3 signaling pathway
EMT plays a key role in cancer progression and metastasis across numerous malignancies, and its regulation is closely linked to the STAT3 signaling pathway. In CRC, HOXB8 promotes proliferation, invasion, and EMT through STAT3 activation, while HOXB8 knockdown suppresses tumor growth and reduces EMT marker expression (81). In PDAC, interleukin-6 (IL-6) secreted by PSCs induces EMT via the Stat3/Nrf2 pathway, thereby promoting migration and invasion; inhibition of either pathway significantly reduces these effects (82). In ovarian cancer, the traditional Chinese medicine Guizhi-Fuling Wan (GZFL) suppresses tumor growth and EMT by downregulating STAT3 activity, supporting its potential as a therapeutic agent (83). In triple-negative breast cancer (TNBC), the natural compound bufotalin induces apoptosis, inhibits proliferation, and reduces metastasis by targeting STAT3 and modulating EMT markers and matrix remodeling enzymes (84). In PC, combining gemcitabine with the STAT3 inhibitor Stattic enhances cytotoxicity by inducing oxidative stress, mitochondrial apoptosis, and DNA damage, while concurrently reducing EMT and immune evasion through downregulation of PD-L1 and CD47 and inhibition of the Smad2/3 pathway (85). This combination therapy also affects broader signaling networks, including those involving the AKT and β-catenin pathways. These findings highlight the central role of STAT3 in regulating EMT, metastasis, and therapeutic resistance, underscoring its value as a comprehensive therapeutic target across multiple cancer types. TNF-α-inducing protein (Tipα), a recently identified carcinogenic factor secreted by Helicobacter pylori (H. pylori), is recognized as a potent inducer of EMT, a key process in cancer cell migration. However, its precise molecular mechanism remains unclear. Growing evidence suggests that aberrant activation of the oncogenic transcription factor STAT3 is a common feature across various types of cancer, including gastric cancer. In SGC7901 gastric cancer cells, Tipα treatment significantly reduced E-cadherin expression while increasing N-cadherin and vimentin expression, accompanied by morphological changes characteristic of EMT. Tipα also enhanced cellular proliferation and migration. Mechanistically, Tipα activates the interleukin-6 (IL-6)/STAT3 signaling pathway, whereas inhibition of this axis reverses Tipα-induced proliferation, migration, and EMT, indicating that Tipα's carcinogenic effects are mediated by IL-6/STAT3 signaling (86).
Lung cancer, the most prevalent cancer type, has a complex and poorly understood pathophysiology, leading to limited therapeutic options and poor prognosis. To clarify the molecular mechanisms driving lung cancer progression, the role of SH2B3 was investigated. SH2B3 expression was significantly reduced in lung cancer tissues and cell lines, whereas TGF-β1 levels were elevated. Low SH2B3 expression was associated with poorer patient outcomes. Functional studies revealed that SH2B3 overexpression suppressed anoikis resistance, proliferation, migration, invasion, and EMT in lung cancer cells, whereas TGF-β1 promoted these malignant traits by downregulating SH2B3. Mechanistically, SH2B3 interacted with JAK2 and SHP2, thereby inhibiting the JAK2/STAT3 and SHP2/Grb2/PI3K/AKT pathways, ultimately reducing EMT and other malignant properties. In vivo experiments confirmed that SH2B3 overexpression significantly restrained lung cancer growth and metastasis. Together, these findings identify SH2B3 as a tumor suppressor in lung cancer that counteracts oncogenic signaling and cellular processes associated with disease progression (87).
The progression and metastasis of CRC and lung cancer involve complex EMT and MET processes tightly regulated by STAT3 signaling. In CRC, STAT3 knockdown increases E-cadherin expression and decreases mesenchymal markers such as N-cadherin and vimentin, thereby reducing invasiveness and enhancing chemosensitivity to fluorouracil. Conversely, STAT3 overexpression promotes EMT and strengthens invasive capacity. ZEB1, a key EMT transcription factor, is upregulated by STAT3 and represses E-cadherin, thereby mediating STAT3-driven invasion. Notably, the expression of both STAT3 and ZEB1 is strongly correlated with advanced TNM stages.
In lung cancer, bone marrow-derived mesenchymal stem cells (BMSCs) under hypoxic conditions release exosomes containing miR-193a-3p, miR-210-3p, and miR-5100, which activate STAT3 signaling in neighboring cancer cells, inducing EMT and enhancing invasiveness. Among these, miR-193a-3p shows promise as a noninvasive biomarker for diagnosing lung cancer and assessing metastatic potential. Systems biology analyses further reveal that BMSC-derived factors such as LIF and IL-6 selectively phosphorylate STAT3 at serine 727 and tyrosine 705, respectively, thereby regulating EMT-MET transitions and CSC dynamics during early dissemination and pre-metastatic niche formation. Collectively, STAT3 acts as a central regulator of EMT/MET plasticity in CRC and lung cancer, with both direct transcriptional control (via ZEB1) and exosomal miRNA-mediated activation driving tumor invasion, metastasis, and therapy resistance (88-90).
4.3 Wnt/β-catenin signaling pathway
The anticancer potential of garcinol, a natural compound derived from Garcinia indica, has been the focus of extensive investigation. Previous studies demonstrated its ability to preferentially induce apoptosis in breast cancer cells by blocking the NF-κB signaling pathway. Garcinol has been shown to reverse EMT and induce MET in aggressive TNBC cell lines, MDA-MB-231 and BT-549. This phenotypic switch was characterized by increased expression of the epithelial marker E-cadherin and decreased levels of mesenchymal markers, including vimentin, ZEB-1, and ZEB-2. Garcinol also upregulated members of the miR-200 and let-7 families, offering a molecular explanation for MET induction. Significantly, ectopic expression of the NF-κB p65 subunit reduced garcinol-induced apoptosis by converting MET back to EMT, while p65 overexpression and miR-200 inhibition counteracted garcinol’s suppression of cell invasion. Additionally, garcinol treatment promoted β-catenin phosphorylation and inhibited its nuclear localization. These findings were validated in a xenograft mouse model, where garcinol inhibited NF-κB activity, altered miRNA expression, and reduced both vimentin and nuclear β-catenin levels (91).
HCC is one of the most lethal cancers, yet its core genetic mechanisms remain incompletely understood. Genome-wide methylation profiling has identified CLDN3 as an epigenetically regulated gene in cancer. In human HCC, CLDN3 downregulation was detected in 87 of 114 (76.3%) primary tumor samples and was significantly correlated with reduced patient survival (P = 0.021). Multivariate Cox regression further confirmed CLDN3 as an independent prognostic indicator for overall survival (P = 0.014). Loss of CLDN3 expression was observed in 67% of HCC cell lines and was strongly linked to promoter hypermethylation. Restoration of CLDN3 expression in HCC cells suppressed motility, invasiveness, and tumorigenic potential in nude mice. Mechanistically, CLDN3 inhibited metastasis by blocking the Wnt/β-catenin-driven EMT pathway by downregulating key regulators, including GSK3B, CTNNB1, SNAI2, and CDH2 (92).
The Wnt/β-catenin signaling pathway is a key regulator of EMT, metastasis, and tumor progression in multiple cancers, including CRC, NSCLC, and gastric cancer. In CRC, RUNX1 is upregulated, promoting EMT and metastasis by directly interacting with β-catenin and enhancing KIT transcription, thereby activating the Wnt/β-catenin pathway. Notably, RUNX1 itself is also regulated by the Wnt/β-catenin signaling pathway, establishing a feedback loop that sustains tumor progression (93). In NSCLC, elevated FOXP3 expression is associated with poor patient prognosis. It promotes tumor proliferation, EMT, and invasion by directly interacting with β-catenin and TCF4, thereby enhancing the transcription of Wnt target genes, including c-Myc and Cyclin D1 (94). In CRC, TM4SF1 is highly expressed and promotes cancer stemness and EMT via the Wnt/β-catenin/c-Myc/Sox2 pathway, while its downregulation significantly suppresses metastasis and tumorigenesis (95). The importance of Wnt signaling within the TME is evident, as stromal Wnt activity, particularly in cancer-associated fibroblasts (CAFs), exerts variable effects on tumor progression. Wnt antagonists such as Sfrp1 suppress tumor growth and EMT by modulating stromal signaling, while leaving tumor-intrinsic Wnt activity unaffected (96). Different CAF subtypes, influenced by Wnt activity, include myofibroblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs), which play opposing roles: myCAFs suppress EMT, while iCAFs promote it. These findings underscore the multifaceted role of Wnt/β-catenin signaling in driving EMT and metastasis through both tumor-intrinsic mechanisms and microenvironmental interactions across diverse cancers. Recent research has further identified NANOGP8, KIAA1199, IQGAP1, and DNER as key facilitators of cancer progression, primarily through pathways linked to Wnt/β-catenin signaling and EMT. Notably, NANOGP8, rather than NANOG1, is the dominant contributor to NANOG expression in sphere-forming cancer stem-like cells (CSLCs) in gastric cancer (97). It strongly promotes stemness, EMT, metastasis, chemoresistance, and Wnt signaling activity, positioning it as a key oncogenic regulator and a promising therapeutic target. KIAA1199 is significantly upregulated in gastric cancer, where it correlates with poor prognosis and drives proliferation, invasion, and EMT through activation of Wnt/β-catenin signaling and MMPs (98).
In PDAC, IQGAP1 acts as an oncogenic scaffold protein that promotes proliferation, migration, invasion, and EMT by increasing Dishevelled2 (DVL2) expression, enhancing β-catenin nuclear translocation and transcriptional activity, and directly binding to both DVL2 and β-catenin; silencing DVL2 attenuates these effects (99).DNER is highly expressed in breast cancer, especially in triple-negative subtypes, where it correlates positively with β-catenin expression and drives EMT, proliferation, metastasis, and resistance to epirubicin-induced apoptosis through the Wnt/β-catenin pathway. Collectively, these findings underscore the oncogenic roles of NANOGP8, KIAA1199, IQGAP1, and DNER across multiple cancer types, all of which converge on the Wnt/β-catenin signaling pathway to promote malignant transformation, thereby highlighting their potential as molecular targets for future cancer therapies. Elevated Wnt signaling has been increasingly associated with the initiation and progression of human breast cancer. Activation of the canonical Wnt pathway leads to the formation of a β-catenin-T-cell factor (TCF) transcriptional complex, which is thought to drive EMT, a critical process underlying cancer tissue invasion. However, the precise molecular mechanisms by which the β-catenin-TCF complex induces EMT-like changes have remained unclear. Recent studies show that canonical Wnt signaling promotes tumor cell dedifferentiation and invasive behavior through an Axin2-dependent pathway that stabilizes Snail1, a zinc-finger transcription factor central to both developmental and cancer-associated EMT. Axin2 functions as a nucleocytoplasmic chaperone for GSK3β, the key kinase regulating Snail1 protein stability and activity. The widespread dysregulation of Wnt signaling across cancers underscores the importance of the β-catenin-TCF-Axin2-GSK3β-Snail1 signaling axis, providing critical mechanistic insight into EMT regulation in tumor progression (100).
PC is among the deadliest malignancies, marked by early metastasis and a high mortality rate. Subunits of the SWI/SNF chromatin-remodeling complex are recognized as key regulators of tumor development; however, the role of SMARCAD1, a member of this family, in PC remains unclear. Analysis of GEO datasets and immunohistochemical evaluation of patient-derived PC tissues revealed elevated SMARCAD1 expression in tumors, with higher levels correlating with poorer patient survival. Functional studies showed that SMARCAD1 enhances the proliferation, migration, and invasion of PCCs. Mechanistically, SMARCAD1 promotes EMT by activating the Wnt/β-catenin signaling pathway in PC (101).
Extensive studies highlight the central role of the Wnt/β-catenin signaling pathway in driving EMT, stemness, metastasis, and chemoresistance across various cancers, including gastric, colorectal, TSCC, and breast cancers. In gastric cancer, VGLL4 functions as a tumor suppressor by inhibiting EMT through negative regulation of the Wnt/β-catenin signaling pathway; its downregulation promotes proliferation, migration, invasion, and apoptosis resistance (102). DVL3 is highly expressed in CRC and is associated with a poor prognosis, promoting EMT, CSLC traits, and multidrug resistance by activating the Wnt/β-catenin/c-Myc/Sox2 pathway. Silencing DVL3 suppresses these malignant properties in both in vitro and in vivo models (103). In cisplatin-resistant TSCC, SOX8 is upregulated, promoting EMT and stem-like properties by directly activating FZD7, which in turn triggers Wnt/β-catenin signaling (104). Silencing SOX8 reduces chemoresistance and CSC-like features, effects that can be partially rescued by β-catenin overexpression. In breast cancer, especially in basal-like subtypes, Wnt signaling is similarly linked to stemness, EMT, and metastatic potential (105). In an orthotopic breast cancer model, inhibition of Wnt signaling through LRP6 suppression reduced self-renewal and metastatic spread, while restoring epithelial marker expression and downregulating EMT-related transcription factors, such as SLUG and TWIST. Collectively, these findings underscore the pivotal role of Wnt/β-catenin signaling in orchestrating EMT, cancer stemness, metastasis, and therapy resistance across various malignancies, positioning it as a critical target for therapeutic intervention.
Extensive research highlights the crucial role of the Wnt/β-catenin signaling pathway in driving EMT, metastasis, and tumor progression across various cancers, including gastric, ovarian, colorectal, and pancreatic malignancies. In gastric cancer, EphA2 is frequently overexpressed and is strongly correlated with EMT markers and metastatic potential, promoting cell migration and invasion by activating the Wnt/β-catenin pathway; this effect can be reversed by pharmacological inhibition of the Wnt/β-catenin pathway. In epithelial ovarian cancer, the tumor suppressor TET1 is notably downregulated in advanced stages of the disease. Restoration of TET1 expression suppresses EMT and tumor cell invasion by demethylating and reactivating Wnt pathway antagonists such as DKK1 and SFRP2, thereby effectively blocking β-catenin signaling (106). TUSC3 is upregulated in CRC tissues and drives EMT, proliferation, invasion, and xenograft tumor growth by activating the MAPK, PI3K/Akt, and Wnt/β-catenin signaling pathways, underscoring its oncogenic role (107). Together, these findings demonstrate that both oncogenic drivers and tumor-suppressive regulators converge on the Wnt/β-catenin pathway to modulate EMT and metastasis, underscoring its therapeutic value across multiple cancers.
EMT is a tightly regulated biological process usually involved in embryonic development and tissue repair, but it becomes dysregulated as cancer progresses. Traditionally, EMT has been viewed as a linear shift from an epithelial to a fully mesenchymal phenotype. However, growing evidence highlights the existence of intermediate or partial EMT states, in which cells acquire mesenchymal traits while still retaining epithelial characteristics. The deubiquitinase USP7 has emerged as a key regulator of this partial EMT phenotype in colon cancer cells. USP7 is markedly overexpressed in colon adenocarcinomas, with its expression correlating with advanced tumor stages. Inhibition or silencing of USP7 significantly reduces mesenchymal marker expression and impairs cancer cell migration. Proteomic analyses have identified the DEAD-box RNA helicase DDX3X as an interacting partner of USP7. Mechanistically, USP7 enhances Wnt/β-catenin signaling by stabilizing DDX3X, thereby promoting invasiveness in CRC cells (103).
Receptor-interacting protein kinase 1 (RIP1), a key mediator of TNF-α signaling, has recently been identified as a novel regulator of canonical WNT/β-catenin signaling, contributing to CRC metastasis. WNT3A treatment progressively increases RIP1 and β-catenin expression, as demonstrated by immunohistochemical analyses of human CRC tissues. These analyses reveal that elevated RIP1 levels correlate with β-catenin expression, tumorigenesis, and metastatic progression. In vivo studies using intravenously injected RIP1-overexpressing CRC cells further demonstrate that RIP1 enhances CRC cell metastatic potential. Mechanistically, WNT3A stimulation promotes direct interaction between RIP1 and β-catenin, stabilizing β-catenin through RIP1-mediated deubiquitination, a process facilitated by suppression of the E3 ligases cIAP1 and cIAP2. Inhibition of cIAP1/2 expression or its ligase activity enhances WNT3A-driven RIP1-β-catenin binding and accumulation, thereby promoting EMT and increasing CRC cell motility and invasion in vitro. Additional experiments confirm the direct interaction between RIP1 and β-catenin, showing that RIP1 disrupts the β-catenin-β-TrCP complex. Collectively, these findings uncover a novel WNT3A-RIP1-β-catenin axis that drives EMT and plays a pivotal role in CRC progression and metastasis (108).
EMT is a key driver of cancer metastasis and therapeutic resistance, orchestrated by intricate regulatory networks, including the Wnt/β-catenin signaling pathway. Computational models suggest that coupled feedback loops between ERK and Wnt signaling tightly regulate E-cadherin expression, with RKIP phosphorylation and transcriptional repression serving as pivotal regulators of the switch-like dynamics of EMT. As a well-characterized EMT suppressor, RKIP loss or downregulation is closely linked to increased metastatic potential (109). In breast cancer, PLAGL2 drives adriamycin resistance and EMT by directly upregulating Wnt6 transcription, thereby amplifying Wnt/β-catenin signaling and promoting proliferation, migration, and invasion. Conversely, PLAGL2 silencing restores adriamycin chemosensitivity (110). RCC2, an oncogenic driver in breast cancer, enhances proliferation and migration through Wnt-mediated EMT, with its overexpression linked to unfavorable patient survival (111). Likewise, CCT5 is overexpressed in lymph node-metastatic gastric cancer, where it promotes proliferation, anoikis resistance, and metastasis by binding to the cytoplasmic domain of E-cadherin, thereby disrupting its interaction with β-catenin and activating Wnt/β-catenin signaling (112).
In summary, these studies collectively demonstrate that oncogenic regulators, such as RKIP, PLAGL2, RCC2, and CCT5, drive EMT and cancer progression by modulating the Wnt/β-catenin axis, underscoring this pathway’s central role in tumor aggressiveness and therapy resistance across various malignancies. EMT, a key determinant of cancer metastasis, stemness, and treatment resistance, is tightly orchestrated by multiple signaling pathways, including the Wnt/β-catenin pathway. In colon cancer, intratumoral heterogeneity in Wnt activity gives rise to diverse EMT states, with the transcription factor RUNX2 identified as a critical epigenetic regulator that remodels chromatin, activates EMT-related genes, and amplifies EMT heterogeneity, an effect strongly associated with poor prognosis across cancer types (113). In laryngeal squamous cell carcinoma, SOX2 overexpression drives migration, invasion, and EMT, characterized by reduced E-cadherin and increased mesenchymal markers, primarily through β-catenin activation (114). In PCa and castration-resistant PCa (CRPC), the nuclear receptor NURR1 promotes EMT, stemness, and metastatic progression by directly activating CTNNB1 and the Wnt/β-catenin pathway, establishing NURR1 as a potential therapeutic target (115). In gastric cancer, RUNX3 is frequently downregulated and inversely correlated with c-MET expression, a known oncogenic driver. Treatment with the c-MET inhibitor INC280 significantly suppresses proliferation, induces apoptosis, and inhibits Wnt signaling and SNAIL expression in c-MET-amplified, RUNX3-positive diffuse-type gastric cancer cells, underscoring its therapeutic potential (116).
Collectively, these findings underscore the central role of Wnt/β-catenin signaling and its transcriptional regulators, RUNX2, SOX2, NURR1, and RUNX3, in driving EMT, metastasis, and therapy resistance across various cancers, highlighting their potential as targets for more effective cancer treatments. STMN2, a key regulator of microtubule disassembly and dynamics, has recently been implicated in cancer progression; however, its role in PDAC remains largely unexplored. In this study, STMN2 was significantly overexpressed in 81 PC tissue samples compared with adjacent normal tissues (54.3% vs. 18.5%; P < 0.01). High expression of STMN2 correlated with aggressive clinical features, including larger tumor size, advanced T stage, lymph node metastasis, and poor prognosis. Elevated STMN2 levels were also strongly associated with cytoplasmic and nuclear β-catenin expression in PCa tissues and cell lines. Functionally, STMN2 overexpression promoted EMT and proliferation in vitro, as evidenced by EMT-like morphological changes, increased motility, modulation of EMT markers (Snail1, E-cadherin, Vimentin), and activation of Cyclin D1 signaling. Pharmacologic inhibition of Wnt/β-catenin signaling using XAV939 attenuated the effects of STMN2 overexpression, while pathway activation with KY19382 restored EMT and proliferation in STMN2-silenced cells. Co-localization of active STMN2 with β-catenin was observed in both cytoplasm and nucleus, and transcriptional regulation of STMN2 by β-catenin/TCF was confirmed through specific binding sites (TTCAAAG). In vivo experiments further demonstrated that STMN2 drives tumor growth by inducing EMT and activating Cyclin D1. Collectively, these findings identify STMN2 as a facilitator of PDAC progression and aggressiveness by promoting EMT and proliferation via a β-catenin/TCF-dependent mechanism (117).
Cervical cancer ranks as the third most common malignancy and the fourth leading cause of cancer-related deaths among women worldwide. In recent years, EMT has attracted increasing attention for its critical role in tumor metastasis. Cysteine-rich intestinal protein 1 (CRIP1) exhibits variable expression across different human cancers; however, its role in cervical cancer remains unclear. Initial findings revealed significantly higher CRIP1 expression in CC tissues compared with adjacent noncancerous tissues. This was further confirmed through qPCR and western blot analyses, which demonstrated elevated CRIP1 levels in CC cell lines. Functional assays using CRIP1 overexpression and siRNA knockdown showed that CRIP1 markedly enhances cell motility and invasion in vitro (P < 0.01). Mechanistic studies indicated that CRIP1 regulates EMT, as reflected by changes in EMT marker expression. Immunohistochemical analysis further revealed significant co-expression of CRIP1 and β-catenin in cervical cancer tissues (P < 0.01). Additionally, CRIP1 upregulated c-Myc, Cyclin D1, and cytoplasmic β-catenin, implicating it in the activation of the Wnt/β-catenin signaling pathway (118).
Extensive research across multiple cancer types highlights the critical role of Wnt/β-catenin signaling in regulating EMT, tumor progression, metastasis, and clinical outcomes. In TNBC, NuSAP1 is markedly overexpressed and associated with poor survival, driving cell proliferation and invasion by activating the Wnt/β-catenin pathway and inducing EMT markers, including Cyclin D1, vimentin, Slug, and Twist (113). Similarly, Hsp90ab1 is upregulated in gastric cancer, where it promotes proliferation and metastasis by binding to and stabilizing LRP5, thereby enhancing Wnt/β-catenin signaling and driving the upregulation of mesenchymal markers (119). Conversely, EFEMP2 acts as a tumor suppressor in bladder cancer; reduced expression correlates with poor prognosis, advanced stage, and grade, as well as with increased EMT and Wnt/β-catenin activity. Its overexpression inhibits proliferation and metastasis (120). In epithelial ovarian cancer, GOLPH3 functions as an oncogene, driving EMT and metastasis via Wnt/β-catenin signaling and upregulating N-cadherin, Snail, cyclin D1, and c-Myc, with EDD identified as a potential downstream effector (121). Pharmacological modulation of Wnt/β-catenin signaling, using activators such as LiCl or inhibitors like XAV939, underscores the central role of this pathway in regulating EMT. Collectively, these findings identify Wnt/β-catenin as a key oncogenic driver linking EMT to cancer progression in TNBC, gastric cancer, bladder cancer, and epithelial ovarian cancer, while also highlighting NuSAP1, Hsp90ab1, EFEMP2, GOLPH3, and EDD as critical regulators and potential therapeutic targets.
4.4. TGF-β signaling pathway
Transforming growth factor-β (TGF-β) plays a complex, context-dependent role in cancer, acting as both a tumor suppressor and promoter. In early-stage tumors, TGF-β can inhibit proliferation; however, some CRC cells evade this control by downregulating TGF-β receptors and disrupting Smad signaling, including Smad4 nuclear translocation. At later stages, TGF-β promotes tumor progression by driving stromal remodeling, angiogenesis, and immune suppression. Secreted in a latent form by cancer cells, TGF-β is activated within the TME by regulatory T (Treg) cells expressing αvβ8 integrin, thereby suppressing cytotoxic T cell activity and facilitating tumor growth. In addition, TGF-β signaling intersects with p53 pathways by repressing REGγ, a proteasome activator that promotes the degradation of tumor suppressors, including p53 and p21. In cancers with mutant p53, this regulation is lost, leading to elevated REGγ expression, increased proteasome activity, and enhanced tumorigenesis and therapy resistance. Collectively, these insights highlight TGF-β’s dual function in cancer, both modulating the intrinsic behavior of tumor cells and shaping an immunosuppressive microenvironment through its signaling and proteolytic interactions (122-124). TGF-β plays a paradoxical, context-dependent role in cancer, functioning both as a tumor suppressor and a promoter of cancer. In the early stages of tumorigenesis, TGF-β acts as a cytostatic and pro-apoptotic factor, creating a crucial barrier against malignant transformation, most evident in PTEN-deficient prostate tumors, where TGF-β signaling restrains progression until overcome by modulators such as COUP-TFII, which blocks SMAD4-dependent transcription and drives metastasis. As tumors evolve, however, TGF-β signaling is frequently co-opted to promote immune evasion, stromal remodeling, and angiogenesis. Tumor cells often upregulate TGF-β to suppress both adaptive and innate immune responses and to activate CAFs, thereby generating an immunosuppressive microenvironment. This reprogramming aligns with a distinct ECM transcriptional profile linked to poor prognosis and resistance to immunotherapies, including PD-1 inhibitors.
Moreover, genetic alterations in key regulators, such as TP53, SMAD4, and MYC, further intersect with TGF-β pathways, thereby amplifying tumor aggressiveness. These insights have sparked growing interest in therapeutic strategies that aim to inhibit TGF-β signaling, thereby restoring immune surveillance and enhancing the efficacy of immunotherapy across various cancer types (125-128). Initially, TGF-β suppresses carcinogenesis by inhibiting cell proliferation and inducing apoptosis; however, as tumors progress, cancer cells and the surrounding stroma hijack TGF-β signaling to drive immune evasion, angiogenesis, and metastasis. Within the TME, CAFs are significant sources of TGF-β, dampening cytotoxic and T helper 1 (TH1) immune responses while fostering a fibrotic, immunosuppressive barrier that limits the effectiveness of immune checkpoint therapies such as PD-L1 blockade. Recent studies suggest that inhibiting TGF-β signaling, particularly in CD4+ T cells using agents like the CD4 TGF-β Trap (4T-Trap), can remodel the TME by enhancing TH2-mediated immunity, normalizing vasculature, and promoting tumor cell apoptosis.
Furthermore, combining TGF-β inhibition with VEGF blockade or checkpoint inhibitors has demonstrated synergistic benefits amplifying anti-tumor immunity and enhancing therapeutic response. Together, these insights highlight the central yet complex role of TGF-β in tumor progression and immune regulation, rendering it a compelling yet challenging target for cancer therapy (129-132). Epithelial and hematopoietic progenitor cells are particularly susceptible to transformation due to their high turnover rates and vulnerability to genetic and epigenetic alterations, including dysregulated responses to growth factors such as TGF-β. TGF-β signaling plays a dual role in cancer, acting as a tumor suppressor in some contexts while promoting tumor progression in others through its complex autocrine and paracrine effects on both tumor cells and the surrounding microenvironment (133). The immune system combats cancer through type 1 immunity, which directly eradicates malignant cells, and type 2 immunity, which supports tissue repair and containment. Loss of TGF-β receptor 2 in CD4+ T cells initiates an IL-4-dependent type 2 response that remodels vasculature, induces hypoxia in cancer cells, and suppresses tumor growth through host-mediated mechanisms (134).
Hypoxia is a critical driver of EMT, invasion, and metastasis in solid tumors. Recent studies have highlighted the role of alternative splicing (AS) in this process, particularly in breast cancer, where hypoxia-induced suppression of the splicing regulator ESRP1 leads to the expression of the pro-metastatic hMENAΔ11a isoform (135). This process is driven by hypoxia-activated TGF-β signaling, which increases the expression of SLUG and RBFOX2, transcriptional repressors of ESRP1. Additionally, RBFOX2 physically interacts with SLUG. Under hypoxic conditions, exosomal TGF-β release amplifies this cascade, inducing broad changes in alternative splicing that promote EMT (136). In lung cancer, TGF-β1-mediated EMT involves the upregulation of neuropilin-2 (NRP2), a receptor for the tumor suppressor SEMA3F, through a TβRI-dependent but SMAD-independent mechanism that engages the ERK and AKT pathways. Elevated NRP2 promotes tumor cell migration, invasion, and morphological changes, which correlate with E-cadherin loss and higher tumor grade. In TNBC, ICAM1 plays a crucial role in bone metastasis by inducing EMT via the TGF-β/SMAD signaling pathway, a process facilitated by integrin interactions (137). High ICAM1 expression correlates with poor prognosis, whereas its inhibition reduces bone metastasis in vivo. In CRC, TGF-β exerts dual functions as both a tumor suppressor and promoter, which are modulated by NDRG2, a gene induced by TGF-β through Sp1 activation and the relief of c-Myc/Miz-1 repression (138). Although NDRG2 normally inhibits EMT and cell invasion, its silencing through promoter hypermethylation weakens the growth-suppressive effects of TGF-β, ultimately facilitating metastasis. Overall, these findings highlight TGF-β as a key regulator of EMT and metastatic progression in various cancers, with factors such as hypoxia, splicing regulators, and epigenetic modifications shaping its dual role as either a tumor suppressor or promoter, depending on the cellular context. Histone methylation is a crucial regulatory mechanism in both normal cellular function and disease, especially in cancer progression. In TGF-β-induced epithelial-mesenchymal transition (EMT), the Polycomb repressive complex 2 (PRC2) component EED, responsible for methylating histone H3 at lysine 27 (H3K27), acts as an essential modulator. When TGF-β initiates EMT, EED expression rises, and its absence interferes with the typical morphological transformations associated with EMT. EED inhibition also counteracts TGF-β-mediated alterations in the expression of essential EMT-related genes, including CDH1, ZEB1, ZEB2, and the miRNA-200 (miR-200) family. Chromatin immunoprecipitation studies further demonstrate that EED contributes to TGF-β-induced transcriptional repression of CDH1 and miR-200 by modulating H3K27 methylation and influencing EZH2 binding at their respective regulatory regions (139).
AlkB homolog 5 (ALKBH5), an RNA N6-methyladenosine (m6A) demethylase, is a critical regulator of development and disease. The role of TGF-β in driving EMT and metastasis in NSCLC, however, remains incompletely understood. In metastatic NSCLC, ALKBH5 expression is markedly reduced. Functionally, ALKBH5 overexpression suppresses TGF-β-induced EMT and invasion in NSCLC cells, while its knockdown enhances these malignant traits. In vivo, ALKBH5 overexpression also limits TGF-β-driven metastasis. Mechanistically, ALKBH5 reduces TGFβR2 and SMAD3 expression and mRNA stability, while increasing SMAD6 levels; knockdown produces the opposite effects. ALKBH5 also influences SMAD3 phosphorylation, reducing SMAD3 activation upon ALKBH5 overexpression and enhancing it upon ALKBH5 silencing. The m6A-binding proteins YTHDF1 and YTHDF3 boost the expression of TGFβR2 and SMAD3, while YTHDF2 inhibits SMAD6 expression. Together, these actions strengthen TGF-β-driven epithelial-mesenchymal transition (EMT) and cell invasion. By removing m6A marks, ALKBH5 destabilizes YTHDF1/3-associated TGFβR2 and SMAD3 transcripts but simultaneously stabilizes SMAD6 mRNA by preventing its degradation through YTHDF2. These findings establish ALKBH5 as a negative regulator of TGF-β-induced EMT and metastasis in NSCLC, operating through a YTHDF1/2/3-dependent mechanism (140).
TGF-β, particularly TGF-β1, plays a central role in driving EMT, tumor progression, and metastasis across multiple cancers by acting on both tumor cells and the surrounding microenvironment. In breast cancer, TGF-β1-induced EMT primes mammary tumor cells for dendritic cell-like migration through the lymphatic system via CCR7/CCL21 signaling. This process is reinforced by p38 MAP kinase and the transcription factor JunB, which enhance CCR7 expression, while TGF-β1 simultaneously upregulates CCL21 in lymphatic endothelial cells, promoting chemotactic migration (141). In bladder cancer, stromal fibroblast-derived TGF-β1, particularly from CAFs, drives EMT and enhances migration and invasion through the FAP/VCAN axis, with VCAN exerting its effects via activation of the PI3K/AKT1 pathway (142). In PCa, TGF-β signaling orchestrates ECM remodeling and desmoplasia within patient-derived tumoroids, thereby promoting epithelial-mesenchymal transition and tumor progression (143). In gastric cancer, the transcription factor SALL4 promotes motility, migration, and invasion by directly activating TGF-β1 transcription, thereby initiating the TGF-β/SMAD pathway and inducing EMT (144); silencing TGF-β1 abolishes these effects. Collectively, these findings highlight the intricate and conserved role of TGF-β1 signaling in regulating EMT, cellular plasticity, and metastatic potential through both tumor-intrinsic mechanisms and interactions with the microenvironment, underscoring its therapeutic value across multiple cancer types. The impact of mesenchymal stem cells (MSCs) on lung cancer progression remains controversial, and the mechanisms underlying this impact remain unclear, limiting their therapeutic application. A previous study revealed that conditioned medium from human umbilical cord MSCs (MSC-CM) promotes EMT, invasion, and migration of lung cancer cells, while concurrently inhibiting their proliferation and inducing apoptosis. The EMT-enhancing effect was attributed to MSC-derived exosomes (MSC-exo), as blocking exosome release abolished this activity. Notably, silencing TGF-β1 expression in MSCs reversed the EMT-inducing effect and enhanced the anti-proliferative and pro-apoptotic functions of MSC-exo in lung cancer cells. Further analysis revealed that MSC-exo activated multiple TGF-β1-associated signaling pathways in lung cancer cells, including the Smad2/3, Akt/GSK-3β/β-catenin, NF-κB, ERK, JNK, and p38 MAPK pathways. The suppression of these pathways upon TGF-β1 silencing highlights MSC-derived TGF-β1 as a key mediator of these effects (145).
Human leukocyte antigen class I (HLA-I) molecules play a pivotal role in immune defense by presenting antigenic peptides to cytotoxic T cells. In both primary and metastatic prostate cancer (PCa), HLA-I expression is frequently reduced, allowing tumor cells to evade immune surveillance and drive disease progression. Elevated levels of growth factors such as TGF-β and epidermal growth factor (EGF) within the TME have been linked to this progression. Notably, both TGF-β and EGF have been shown to suppress HLA-I expression in PCa cells, thereby decreasing their susceptibility to cytotoxic T cell-mediated killing. This reduction in HLA-I is closely linked to classical EMT, as evidenced by accompanying morphological changes and altered gene-expression profiles. Mechanistic studies further identified Snail overexpression as a critical mediator of HLA-I downregulation, with NF-κB/p65 emerging as a potential downstream effector through which Snail exerts its regulatory influence. Collectively, these findings provide the first evidence that TGF-β and EGF can drive HLA-I downregulation in parallel with EMT in PCa cells (146).
TGF-β, particularly TGF-β1, plays a central role in driving tumor progression, invasion, migration, and EMT across multiple malignancies, including gastric, ovarian, lung, and breast cancers. In gastric cancer, TGF-β1 induces EMT by suppressing E-cadherin and elevating vimentin expression, with Krüppel-like factor 8 (KLF8) identified as a key downstream transcription factor mediating this process. Silencing KLF8 reverses EMT traits and reduces cellular motility and invasiveness, highlighting KLF8 as a promising therapeutic target (147). TGF-β1, together with TGF-β2 and TGF-β3, enhances ovarian cancer cell migration without triggering a complete EMT phenotype. This is evidenced by the retention of E-cadherin expression and the preservation of epithelial morphology, suggesting that metastasis may occur through collective cell migration rather than single-cell invasion (148). TGF-β promotes both EMT and the acquisition of stem cell-like traits in lung cancer, likely through epigenetic mechanisms involving DNA demethylation and the activation of genes such as Slug and CD87 (149). In breast cancer,CAFs secrete high levels of TGF-β1, which drive EMT and enhance cancer cell aggressiveness through paracrine activation of the TGF-β/Smad pathway; notably, these effects can be reversed by suppressing TGF-β1 (150). These findings highlight the pivotal and multifaceted role of TGF-β1 signaling in driving cancer progression and EMT, mediated by both intrinsic cellular mechanisms and TME interactions, and underscore the therapeutic promise of targeting key regulators, including KLF8, epigenetic modulators, and TGF-β1 itself.
Signal peptide-CUB-EGF-like domain-containing protein 3 (SCUBE3) is a secreted glycoprotein that is highly expressed in lung cancer tissues and correlates with greater invasive potential in lung cancer cells, implicating it in tumor progression. Functional studies show that exogenous SCUBE3 promotes cell motility and invasion, whereas its knockdown suppresses tumor growth and metastasis in vivo. In the extracellular environment, SCUBE3 is cleaved by the gelatinases MMP-2 and MMP-9, producing two major fragments: the N-terminal EGF-like repeats and the C-terminal CUB (C1r/C1s, Uegf, Bmp1) domain. Both the intact SCUBE3 protein and the isolated CUB fragment bind to the TGF-β type II receptor through the CUB domain, thereby activating TGF-β signaling and inducing EMT. This activation involves phosphorylation of Smad2/3, increased Smad2/3 transcriptional activity, and upregulation of EMT- and progression-related genes, including TGF-β1, MMP-2, MMP-9, plasminogen activator inhibitor-1, VEGF, Snail, and Slug, ultimately driving enhanced migration and invasion of cancer cells (151).
Frizzled receptor 7 (FZD7) is highly expressed in PDAC, where its upregulation is associated with early liver metastasis, enhanced EMT, and increased CSC traits, ultimately contributing to poor clinical outcomes. Mechanistically, FZD7 promotes motility, invasion, and stemness by activating canonical WNT/β-catenin signaling and engaging the TGF-β/SMAD3 pathway. Functional studies demonstrate that FZD7 overexpression enhances the mesenchymal phenotype and increases the expression of CSC markers, including CD24, CD44, and ABCG2, whereas silencing FZD7 reduces EMT, CSC features, and TGF-β1-driven changes. Sphere-forming cells, indicative of stem-like properties, exhibited higher metastatic potential, accompanied by elevated FZD7 expression, underscoring FZD7's key role in promoting hepatic metastasis by coordinating EMT and CSC amplification (152).
TGF-β acts as a key driver of EMT, a central process in tumor metastasis across multiple cancer types. Several mechanisms have been identified through which TGF-β promotes EMT and accelerates cancer progression. In CRC, the fucosyltransferases FUT3 and FUT6 enhance malignancy by fucosylating type I TGF-β receptors, thereby amplifying downstream signaling and promoting EMT-mediated migration and invasion (153). In PC, TGF-β drives EMT through ROS generated by NADPH oxidase 4 (Nox4), with protein tyrosine phosphatase 1B (PTP1B) acting as a redox sensor that transmits EMT signals. Notably, Nox4, TGF-β, and EMT markers, such as N-cadherin, are upregulated in patient tumors (154). In ovarian cancer, TGF-β1 promotes EMT through epigenetic regulation, inducing extensive DNA methylation in CpG islands of EMT-related gene promoters, including CDH1 and COL1A1, leading to gene silencing and enhanced metastasis (155). These effects are driven by the upregulation of DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) but can be reversed using DNMT inhibitors such as SGI-110. In lung cancer, TGF-β1-induced EMT activates the histone acetyltransferases p300/CBP, which acetylate Smad2 and Smad3, thereby enhancing transcription of EMT-associated genes (156). This pathway can be blocked by EGCG, a p300/CBP inhibitor, thereby reversing EMT-related gene expression. Collectively, these findings highlight the intricate regulatory mechanisms that span glycosylation, redox signaling, histone modification, and DNA methylation, through which TGF-β signaling drives EMT and metastasis. They also highlight the therapeutic potential of targeting key mediators, such as FUTs, Nox4, PTP1B, p300/CBP, and DNMTs, to suppress cancer progression.
The role of the cadherin switch, characterized by E-cadherin downregulation and N-cadherin upregulation during EMT in the progression of extrahepatic cholangiocarcinoma (CC), was investigated. In CC cell lines, TGF-β1 induced EMT, resulting in increased motility, invasion, and fibroblast-like morphology, accompanied by a cadherin switch. Analysis of tumor tissues from 38 patients revealed that reduced E-cadherin expression, as well as the combined loss of E-cadherin and gain of N-cadherin, were significantly associated with poorer survival outcomes. These findings suggest that TGF-β-induced EMT and the cadherin switch play critical roles in driving tumor progression in extrahepatic cholangiocarcinoma, highlighting them as potential targets for elucidating mechanisms of invasion and metastasis (157).
IGHG1 was markedly overexpressed in gastric cancer tissues compared with normal tissues and was found to drive proliferation, migration, invasion, and EMT in SGC7901 gastric cancer cells. Proteomic profiling, immunohistochemistry, and Western blot analysis all confirmed elevated IGHG1 levels in tumor samples. Functional assays revealed that IGHG1 knockdown suppressed cell proliferation, migration, and invasion, while altering EMT-related protein expression, specifically decreasing Vimentin, N-cadherin, TGF-β, and phosphorylated SMAD3, and increasing E-cadherin. These results suggest that IGHG1 facilitates EMT in gastric cancer by modulating the TGF-β/SMAD3 signaling pathway (158).
4.5. Notch signaling pathway
The Notch signaling pathway exerts a complex, context-dependent influence on cancer progression and therapeutic response. Initially identified for its role in embryonic development, Notch is now recognized as a key regulator of adult tissue homeostasis and tumorigenesis. Aberrant Notch activation has been linked to cancer initiation, progression, and metastasis, including a recently identified stroma-dependent role in osteolytic bone metastases. Depending on the tumor type and biological context, Notch may function as either an oncogene or a tumor suppressor. In small-cell lung cancer (SCLC), elevated Notch activity is associated with reduced neuroendocrine (NE) differentiation, enhanced antigen presentation, and greater cytotoxic T-cell infiltration, collectively predicting improved responses to immune checkpoint blockade.
Additionally, Notch interacts with central regulatory networks such as p53 and p63, shaping stem cell potential and differentiation, with implications for targeted and combinatorial therapies. Collectively, these findings underscore both the complexity and therapeutic potential of modulating Notch signaling in diverse cancer settings (159-162). The Notch signaling pathway plays a critical, multifaceted role in cancer, strongly associated with tumor progression, therapeutic resistance, and CSC biology. CSCs exploit Notch signaling to sustain self-renewal, differentiation, and survival, thereby driving tumor recurrence and metastasis. In several malignancies, including SCLC, Notch exhibits context-dependent functions, acting as either a tumor suppressor or a tumor promoter, depending on the cellular environment and intratumoral heterogeneity. In SCLC, for instance, Notch activation induces a shift from the NE to a non-NE phenotype via REST, suppressing NE proliferation while simultaneously promoting slow cycling. These chemoresistant cell populations support tumor persistence and immune evasion.
Beyond this, Notch regulates angiogenesis, immune responses, and CSC maintenance, underscoring its dual role in cancer biology. Despite these complexities, therapeutic strategies targeting Notch, particularly when combined with chemotherapy or with modulators of pathways such as Wnt and Hedgehog, hold promise for eliminating both CSCs and bulk tumor cells. However, clinical progress remains hindered by its context-specific activity and extensive pathway crosstalk. A deeper understanding of Notch’s diverse functions and interactions with other signaling networks will be essential for designing targeted and combinatorial treatments to improve cancer outcomes (163-166). The Notch signaling pathway, which regulates cell proliferation, differentiation, and apoptosis in a context-dependent manner, has been identified as a tumor suppressor in bladder cancer. Inactivating mutations in components of the Notch pathway have been detected in more than 40% of human bladder tumors analyzed, underscoring their critical role in carcinogenesis. Unlike previously established driver mutations such as FGFR3 and RAS, Notch activation in bladder cancer cells suppresses proliferation by upregulating dual-specificity phosphatases (DUSPs), which in turn reduce ERK1/2 phosphorylation. In mouse models, loss of Notch signaling results in increased ERK1/2 activity and the development of urinary tract tumors. Collectively, these findings highlight Notch inactivation as a key driver of urothelial carcinoma progression (167).
Notch signaling exhibits diverse, context-dependent roles in cancer, influencing tumor growth, EMT, and therapeutic response. In bladder cancer, loss-of-function mutations in Notch1 and Notch2 drive carcinogenesis and promote the development of squamous cell carcinoma with mesenchymal traits in mouse models (168). The Notch downstream effector HES1 plays a key role in maintaining the epithelial phenotype, and its loss drives EMT and the acquisition of aggressive tumor traits. In CRC, Notch1 signaling functions oncogenically, with higher expression observed in tumor tissues compared with normal colon tissue. This activation promotes EMT and stem-like properties by upregulating CD44, Slug, Smad-3, and Jagged-1, ultimately enhancing migratory capacity and anchorage-independent growth (169). Blocking this pathway with DAPT or Jagged-1-Fc reduced these effects, highlighting its pro-tumorigenic role. In gastric cancer, Notch1 signaling drives tumor progression; luteolin, which suppresses Notch1, reverses EMT and reduces proliferation, invasion, and migration in both in vitro and in vivo models (170).
EMT is a fundamental process in embryonic development, tissue repair, and cancer progression. Cells undergoing a partial EMT, known as a hybrid epithelial/mesenchymal (E/M) state, often migrate collectively and form CTC clusters, which are key drivers of metastasis. Intercellular signaling pathways, such as the Notch pathway, can induce either partial or complete EMT, although the mechanisms governing cluster formation remain unclear. An integrated computational and experimental study revealed that Numb, a negative regulator of Notch signaling, plays a critical role in controlling EMT and cluster formation. Computational modeling indicates that Numb prevents a complete EMT by maintaining cells in a hybrid E/M phenotype. Consistently, silencing Numb in H1975 cells, which generally display a stable hybrid E/M state, induces a complete EMT, confirming its role in stabilizing the phenotype by regulating Notch-driven EMT dynamics. At the multicellular scale, the model suggests that Numb influences the balance between hybrid E/M and fully mesenchymal cells within clusters, thereby enhancing tumor-initiating capacity. Clinically, elevated Numb expression is associated with poor survival outcomes in multiple independent lung and ovarian cancer datasets, underscoring its link to heightened cancer aggressiveness (171).
Notch signaling plays a central role in carcinogenesis, functioning as either an oncogene or a tumor suppressor, depending on the cancer type. Increasing evidence highlights its influence on critical cellular processes, including proliferation, apoptosis, cell cycle regulation, and metastasis. In PCa, Notch-1 has been shown to regulate invasion, migration, and EMT. Overexpression of Notch-1 in PC-3 cells enhances their invasive and migratory abilities, whereas Notch-1 silencing suppresses these traits. Furthermore, high Notch-1 expression drives EMT in PC-3 cells, as reflected by changes in EMT marker profiles and the acquisition of CSC-like properties (172).
PC remains one of the most challenging malignancies to treat, mainly due to both intrinsic and acquired resistance to chemotherapy and radiation. Gemcitabine, the standard therapeutic agent either alone or in combination with other drugs, has provided only limited improvements in overall survival for patients with advanced disease. Previous studies have shown that gemcitabine-resistant PCCs frequently exhibit an EMT phenotype similar to that of CSLCs, although the underlying molecular mechanisms remain poorly defined. Emerging evidence indicates that Notch-2 and its ligand Jagged-1 are markedly upregulated in gemcitabine-resistant cells, consistent with Notch signaling's role in promoting EMT and stem-like properties. Blocking Notch signaling reduces the invasive potential of gemcitabine-resistant cells and partially reverses EMT, inducing MET. This reversal is characterized by the downregulation of EMT markers, including vimentin, ZEB1, Slug, Snail, and NF-κB. Collectively, these findings establish a mechanistic link between heightened Notch activity and the EMT-driven chemoresistant phenotype of PC, suggesting that therapeutic targeting of the Notch pathway could represent an effective strategy to overcome resistance and limit tumor progression and metastasis (173).
EMT plays a key role in driving PCa metastasis, with Notch signaling, particularly Notch-4, closely implicated in this process. In PCa cell lines DU145, PC3, and LnCAP, Notch-4 expression is significantly higher compared to non-malignant prostate epithelial cells. Silencing Notch-4 reduces cell viability and proliferation, most notably in DU145 and PC3 cells, and induces apoptosis in PC3 cells. In addition, Notch-4 inhibition suppresses cell motility and invasion and alters expression of EMT markers. Mechanistically, this effect appears to be mediated by reduced NF-κB activity, as pharmacological activation of NF-κB with PMA reverses the inhibitory effects of Notch-4 silencing on proliferation, apoptosis, and EMT features. These findings suggest that targeting Notch-4 effectively suppresses PCa progression by disrupting the EMT and NF-κB signaling pathways (174). Activation of Notch1 reduces epithelial markers, such as E-cadherin and cytokeratins, while simultaneously increasing the expression of EMT inducers, including Slug and Snail, and leading to marked morphological changes. Notch1 also reinforces TGFβ/Smad signaling by elevating TGFβ and its type I receptor expression, thereby sustaining prolonged Smad phosphorylation. Blocking Notch signaling with DAPT or silencing Notch1 reduces TGFβ-induced EMT features, demonstrating that Notch activity is essential for full TGFβ-driven EMT. Conversely, TGFβ enhances the expression of the Notch ligand Jagged1 and the downstream target HES1, creating a positive feedback loop. Co-activation of Notch1 and TGFβ markedly increases cell motility and migration compared to either pathway alone, highlighting the synergistic role of Notch-TGFβ signaling in regulating EMT and promoting epithelial ovarian cancer progression (175).
4.6. Hedgehog signaling pathway
EMT is a central driver of treatment resistance, metastasis, and recurrence across multiple cancers, with growing evidence indicating that the Hedgehog (Hh) pathway, particularly through the transcription factor Gli1, is a key regulator of these processes. In HNSCC, resistance to EGFR-targeted therapy with cetuximab is associated with elevated Gli1 and vimentin expression, indicative of EMT activation (176). Blocking the Hedgehog (Hh) pathway with IPI-926 reduces cetuximab resistance by shifting cells toward a more EGFR-dependent, epithelial state, thereby enhancing cetuximab’s effectiveness and even achieving complete tumor regression in specific patient-derived xenograft models. Similar trends are observed in lung squamous cell carcinoma, where Gli1 expression inversely correlates with E-cadherin and β-catenin levels; silencing Gli1 suppresses migration and restores epithelial characteristics (177).
In ovarian cancer, activation of the Shh-Gli1 signaling pathway by purmorphamine drives EMT and enhances invasiveness, with contributions from crosstalk between the PI3K-Akt pathway. These findings suggest that simultaneously targeting both pathways may provide an effective therapeutic strategy (178). Studies on PC demonstrate that SHH-Gli1 signaling promotes EMT and metastasis by regulating a broad spectrum of downstream pathways, including TGFβ, Wnt, PI3K/AKT, Ras, and integrins. Gene expression profiling further reveals multiple Gli1-regulated targets, highlighting its central role in tumor progression (179). Together, these studies establish the SHH-Gli1 axis as a key regulator of EMT across multiple tumor types, supporting the inhibition of this axis as a promising strategy to overcome therapy resistance, prevent metastasis, and improve clinical outcomes. Metastatic disease remains the leading cause of breast cancer-related mortality, with limited therapeutic options capable of significantly extending patient survival. Nitidine chloride (NC), a naturally occurring polyphenolic compound, has demonstrated notable anticancer activity in various malignancies, including breast cancer. A study investigated the effects of NC on EMT and the acquisition of CSC-like traits in breast cancer cells, two processes crucial to tumor progression and dissemination, to elucidate the underlying molecular mechanisms.
Using the MDA-MB-468 and MCF-7 cell lines, NC markedly suppressed cancer cell migration and invasion, as confirmed by scratch wound and Transwell assays. NC also reduced mammosphere formation and diminished CSC-like subpopulations, as determined by flow cytometry. Mechanistically, NC downregulated core components of the Hedgehog signaling pathway, Smoothened, Patched, Gli1, and Gli2, leading to decreased expression of EMT-inducing transcription factors such as Snail, Slug, and ZEB1. This corresponded with a reversal of EMT, evidenced by reduced N-cadherin and vimentin levels alongside increased E-cadherin expression. Additionally, NC suppressed CSC markers, including Nanog, Nestin, Oct-4, and CD44, reinforcing its role in limiting stemness. Significantly, NC counteracted TGF-β1-induced EMT and CSC-like features, confirming its ability to curb metastatic potential. Collectively, these findings demonstrate that NC inhibits both EMT and CSC-like properties in breast cancer by underscorblocking Hedgehog signaling, highlighting its potential as a therapeutic candidate for metastatic breast cancer (180).
Hypoxia is a major driver of EMT and invasion in cancer, yet the molecular mechanisms underlying this process remain incompletely defined. Previous studies examined the role of Hedgehog (Hh) signaling in mediating hypoxia-induced EMT and invasion in PC. PCCs were cultured under hypoxic (3% O₂) and normoxic conditions, with interventions including HIF-1α siRNA, the Smoothened (SMO) inhibitor cyclopamine, and GLI1 siRNA to assess the involvement of these pathways. Results showed that hypoxia induces EMT, enhances invasive capacity, and activates a non-canonical, ligand-independent form of Hh signaling that does not alter SHH or PTCH1 expression. Silencing HIF-1α abolished hypoxia-induced EMT and invasion without altering SHH or PTCH1 levels, confirming the non-canonical nature of the pathway. Inhibition of Hh signaling through cyclopamine or GLI1 siRNA reduced EMT and invasion, though only GLI1 knockdown suppressed vimentin expression, a key EMT marker. These findings suggest that under hypoxia, GLI1 activation and subsequent EMT occur independently of SMO, likely through alternative mediators, including TGF-β, KRAS, or RTKs. Collectively, the results indicate that hypoxia accelerates PC progression by engaging a non-canonical Hh signaling route, positioning GLI1 as a central regulator of hypoxia-driven EMT and invasion. Targeting this pathway may represent a promising therapeutic strategy to limit PC metastasis (181).
The Hedgehog (Hh) signaling pathway plays a crucial role in driving EMT, promoting CSC-like traits, and enhancing invasion, metastasis, and chemoresistance across multiple cancers, particularly pancreatic, gastric, and NSCLC. In PC, studies have shown that Hh signaling remains highly active in tumor spheres enriched for CSCs, where it supports self-renewal, induces EMT, and contributes to increased invasiveness, therapeutic resistance, and metastatic potential (182). Blocking key Hedgehog (Hh) pathway components, such as Smoothened (SMO) and GLI1, effectively suppresses malignant behaviors, underscoring the pathway’s critical role in sustaining CSCs and driving tumor progression. GLI1 further amplifies EMT by directly upregulating the EMT-associated gene S100A4, linking Shh-GLI1 signaling to enhanced motility and metastatic potential. In gastric cancer, Shh overexpression correlates with poor prognosis and increased metastasis, primarily through PI3K/Akt-mediated EMT and the induction of MMP-9 activity (183). In non-small cell lung cancer (NSCLC), Hedgehog signaling contributes to resistance against the EGFR tyrosine kinase inhibitor (TKI) erlotinib, especially in cells that have undergone TGF-β1-induced EMT (184). In non-small cell lung cancer (NSCLC), Hedgehog signaling contributes to resistance against the EGFR TKI erlotinib, especially in cells that have undergone TGF-β1-induced epithelial-mesenchymal transition (EMT) (185). These findings highlight Hedgehog (Hh) signaling as a key regulator of EMT, CSC traits, and therapy resistance, suggesting that therapeutic targeting of this pathway, especially at the GLI1 level, holds strong potential for treating multiple aggressive cancer types.
The Sonic Hedgehog (Shh) signaling pathway has emerged as a critical regulator of EMT, CSC traits, tumorigenicity, invasion, and therapy resistance in multiple cancers, including bladder, prostate, gastric, and colorectal malignancies. In bladder cancer, TGF-β1-induced Shh activation drives EMT, characterized by reduced E-cadherin and increased N-cadherin expression, alongside enhanced motility, stemness, and tumorigenic potential. These effects, however, can be effectively counteracted by Hedgehog pathway inhibitors such as cyclopamine and GDC-0449 (186). Similar findings in PCa reveal that Shh and androgen (DHT) signaling synergistically promote EMT and elevate osteonectin expression, particularly in CAFs. Inhibition of Shh signaling reduces these effects, suggesting promising therapeutic potential (187). In colon cancer, CSC-enriched spheres exhibit high expression of stemness and EMT markers, which are significantly diminished by cyclopamine treatment, highlighting the link between Shh signaling and therapy resistance (188). In gastric cancer, activation of the Shh pathway is linked to poor prognosis and increased tumor aggressiveness, with Gli1 overexpression driving EMT by suppressing E-cadherin and upregulating vimentin, ultimately enhancing migration and invasion (189). Inhibition of the Shh/Gli1 pathway with GANT61 reduces these malignant traits. Across studies, Shh signaling is consistently linked to the promotion of EMT and CSC phenotypes, thereby driving metastasis and treatment resistance. Targeting downstream effectors, such as Gli1, has proven effective in reversing EMT, diminishing stemness, and reducing tumor invasiveness, thereby establishing the Shh/Gli1 axis as a promising therapeutic target in multiple solid tumors.
The hypoxic microenvironment, a hallmark of PC, plays a critical role in driving tumor growth, metastasis, and EMT. Natural compounds, such as curcumin, have recently garnered attention for their ability to inhibit PC progression. However, the specific ability of curcumin to counteract hypoxia-induced tumor advancement and the underlying molecular mechanisms remain insufficiently understood. Previous studies investigated the effects of curcumin on hypoxia-induced EMT and Hedgehog (Hh) pathway activation in PC using the human Panc-1 cell line under hypoxic conditions. Cell proliferation was assessed using the MTT assay, whereas migration and invasion were evaluated using wound-healing and Transwell assays. Expression of EMT markers, including E-cadherin, N-cadherin, and vimentin, was analyzed by quantitative PCR, Western blotting, and immunofluorescence. Additionally, the key components of the Hh pathway, including SHH, SMO, and GLI1, were examined using western blot analysis. Results demonstrated that curcumin markedly suppressed hypoxia-induced proliferation, migration, and invasion of PCCs, modulated EMT marker expression, and significantly reduced hypoxia-driven activation of the Hh pathway (190).
The downstream mechanisms by which the SDF-1/CXCR4 axis regulates invasion in PC remain incompletely understood. Emerging evidence indicates that SDF-1/CXCR4 signaling promotes PC progression through its interaction with the non-canonical Hedgehog (Hh) pathway. Activation of CXCR4 by its ligand SDF-1 has been shown to enhance invasion, induce EMT, and activate non-canonical Hh signaling in CXCR4-positive PCCs. Importantly, these invasive and EMT-promoting effects are suppressed by the Smoothened (SMO) inhibitor cyclopamine or by GLI1-targeted siRNA. Collectively, these findings suggest that the SDF-1/CXCR4 axis regulates the noncanonical Hh pathway by ligand-independent upregulation of SMO transcription, thereby driving EMT and invasive behavior in PC (191).
4.7. NF-κB signaling pathway
The transcription factor NF-κB is frequently activated across a wide range of human cancers and promotes carcinogenesis primarily by protecting transformed cells from apoptosis. Using an established in vitro and in vivo mammary carcinogenesis model driven by Ha-Ras and TGF-β, researchers investigated the role of this pathway in epithelial plasticity and metastasis. Results demonstrated that the IKK-2/IκBα/NF-κB signaling cascade is essential for both the initiation and maintenance of EMT. Blocking NF-κB signaling prevented EMT in Ras-transformed epithelial cells. In contrast, activation of NF-κB alone was sufficient to induce a mesenchymal phenotype, even in the absence of TGF-β stimulation.
Furthermore, reducing NF-κB activity in cells that had already undergone EMT led to a reversion toward an epithelial state, underscoring NF-κB’s critical role in sustaining EMT. Consistent with EMT’s contribution to invasion and metastasis, NF-κB inhibition also abolished the metastatic potential of mammary epithelial cells in mouse models. Together, these findings emphasize NF-κB’s pivotal involvement at multiple stages of breast cancer progression and reveal that cooperation between Ras and TGF-β signaling in advanced carcinogenesis is heavily dependent on NF-κB activity (192).
The metastatic progression of breast cancer is closely linked to the expression of EMT-inducing transcription factors (EMT-TFs) such as SNAIL, SLUG, SIP1, and TWIST1. The presence of these EMT-TFs and the activation of nuclear factor-κB (NF-κB) are both associated with increased tumor aggressiveness and metastatic potential. In aggressive breast cancer cell lines MDA-MB-231 and HCC-1954, suppression of NF-κB/p65 activity, either via siRNA-mediated knockdown or treatment with the inhibitor dehydroxymethylepoxyquinomicin (DHMEQ), significantly reduced cell migration and invasion. This inhibition was accompanied by downregulation of SLUG, SIP1, TWIST1, MMP11, and N-cadherin, alongside upregulation of E-cadherin. In contrast, the less aggressive MCF-7 cell line displayed no substantial changes under similar conditions. Bioinformatic analysis identified multiple NF-κB binding motifs within the promoter regions of SNAIL, SLUG, SIP1, and TWIST1. Chromatin immunoprecipitation and luciferase reporter assays further confirmed direct NF-κB/p65 binding to the promoters of TWIST1, SLUG, and SIP1. Collectively, these findings establish NF-κB as a direct transcriptional regulator of EMT-TFs, driving the invasive phenotype of breast cancer, and highlight NF-κB as a promising therapeutic target for mitigating breast cancer metastasis (193).
EMT, a critical process in cancer progression and metastasis, is regulated by multiple signaling pathways and molecular factors, with the transcription factor NF-κB playing a central role in many tumor types. In colon cancer, genistein has been shown to inhibit TNF-α- induced EMT by upregulating E-cadherin and suppressing N-cadherin and key EMT-related transcription factors, including Slug, ZEB1/2, FOXC1/2, and TWIST1 (194). This inhibitory effect is mediated by suppression of the Notch1/NF-κB signaling pathway, highlighting genistein’s potential as an antimetastatic agent. In breast cancer, the VEGF/NRP-1 axis drives proliferation, migration, invasion, and EMT by activating both NF-κB and β-catenin pathways (195). Blocking VEGF or NRP-1 reverses these effects and increases GSK-3β activity, underscoring this pathway as a promising therapeutic target.
Additionally, in breast cancer, chemotherapy-induced EMT is driven by the suppression of miR-448, which restrains SATB1 and amphiregulin expression. Loss of miR-448 consequently enhances EGFR-mediated TWIST1 expression and activates NF-κB signaling (196). A self-reinforcing feedback loop exists in which elevated NF-κB suppresses miR-448 transcription, thereby amplifying EMT and promoting treatment resistance. Clinical samples from patients receiving various chemotherapy regimens revealed reduced miR-448 levels accompanied by increased SATB1 and TWIST1 expression, confirming the clinical relevance of this regulatory axis. In PC, NF-κB remains constitutively active and drives EMT by upregulating mesenchymal markers such as vimentin and ZEB1, while simultaneously downregulating E-cadherin, even in the absence of functional TGF-β signaling (197).
Blocking NF-κB activity prevents EMT induction and reduces invasive capacity, whereas activating NF-κB markedly enhances these malignant traits. Collectively, these findings underscore NF-κB’s central role in driving EMT, invasion, and therapy resistance across multiple cancer types, positioning it as a critical therapeutic target. EMT is a fundamental process in tumor progression and metastasis, tightly regulated by interconnected signaling networks in which NF-κB plays a key role. In breast cancer, matrix metalloproteinase-3 (MMP-3) induces EMT by activating NF-κB via ROS, which in turn directly regulates Snail transcription (198).
In mammary epithelial cells, constitutively active NF-κB (p65) suppresses epithelial markers such as E-cadherin while inducing mesenchymal traits via ZEB1 and ZEB2. This shift reduces acinar formation and promotes a mesenchymal phenotype, with TNFα sustaining the EMT state. In colon cancer, genistein counteracts epithelial-mesenchymal transition and limits cell migration by downregulating the Notch1/NF-κB/Slug/E-cadherin axis. In PC, NF-κB activation plays a central role in driving EMT, metastasis, and neural invasion. Pharmacological inhibition with BAY 11-7085, triptolide, or Minnelide reduces EMT markers (SNAI1, SNAI2, ZEB1), decreases tumor burden and metastatic spread, and restores epithelial features (199). Furthermore, NF-κB contributes to neural invasion by strengthening tumor-nerve interactions through the induction of neurotrophin production and the stimulation of nerve growth. In CD133⁺ PC stem-like cells, the CCL21/CCR7 signaling axis drives epithelial-mesenchymal transition and metastasis via activation of the Erk/NF-κB pathway (200).
The persistent activation of the transcription factor NF-κB plays a crucial role in driving EMT, tumor progression, and therapy resistance across multiple cancers, including breast, lung, and colorectal malignancies. In breast cancer models using MCF10A cells, overexpression of the NF-κB subunit p65 suppresses epithelial markers, including E-cadherin and desmoplakin. In contrast, it upregulates the mesenchymal marker vimentin and the EMT regulators ZEB1 and ZEB2. These changes result in an EMT-like morphology and impaired acinar formation in 3D cultures (201). TNFα can induce a similar phenotype, which is reversible upon TNFα withdrawal, underscoring the dynamic nature of NF-κB-driven EMT. In lung cancer, hypoxia reduces cisplatin sensitivity by promoting EMT and stemness through NF-κB signaling (202). However, 20(R)-ginsenoside Rg3 counteracts this effect by suppressing NF-κB, thereby restoring cisplatin's efficacy both in vitro and in vivo.
In breast cancer, metformin selectively targets epithelial cells that respond to treatment, while reducing mesenchymal marker expression in resistant cells (203). Additionally, it reverses IL-6-induced EMT by inhibiting STAT3 and NF-κB via AMPK activation, thereby reducing proliferation and migration. In CRC, cells overexpressing the NF-YAl splice variant exhibit heightened EMT, decreased adhesion, amoeboid motility, and increased metastatic potential compared to those expressing the less aggressive NF-YAs isoform. These findings establish NF-YAl as both a prognostic indicator and a potential therapeutic target in metastatic CRC (204). Overall, these findings underscore NF-κB as a central regulator of EMT, tumor aggressiveness, and therapy resistance, suggesting that targeting NF-κB and its downstream pathways could offer promising therapeutic strategies to suppress metastasis and improve treatment outcomes.
4.8. HIF-1α signaling pathway
CD133, a well-recognized marker of CSCs, plays a context-dependent role in promoting tumor aggressiveness under hypoxic conditions. Evidence indicates that CD133 regulates HIF-1α production and enhances tumor cell motility. Comparative studies of the CD133-positive PCC line Capan1M9 and the CD133-negative shCD133M9 line under hypoxia revealed markedly elevated HIF-1α expression in Capan1M9, confirmed by Western blotting, nuclear translocation assays, and real-time PCR. Luciferase reporter assays further demonstrated enhanced activation of hypoxia-responsive elements (HREs) in CD133-positive cells. Functionally, these cells exhibited greater migratory ability in wound-healing and migration assays and expressed higher levels of EMT-related genes than their CD133-negative counterparts. Collectively, these findings suggest that elevated HIF-1α in CD133-positive PCCs under hypoxic conditions drives increased migration via EMT gene activation, highlighting the role of CD133 in tumor aggressiveness and its potential as a therapeutic target for eradicating pancreatic CSCs (205).
HIF-1α plays a central role in driving EMT, metastasis, and therapy resistance across multiple cancers, including pancreatic, gastric, prostate, and colorectal malignancies. Both hypoxic conditions and HIF-1α overexpression promote EMT by downregulating epithelial markers, such as E-cadherin, while upregulating mesenchymal markers, including N-cadherin and vimentin, as well as EMT-associated transcription factors, including Twist, Snail, and ZEB1. In PC, HIF-1α-induced EMT is mediated mainly by NF-κB activation, and inhibition of this pathway reverses EMT, reducing both invasiveness and treatment resistance (206). In gastric cancer, HIF-1α drives peritoneal metastasis by activating TGF-β signaling and upregulating MMP-2 and EMT markers. In contrast, dextran sulfate (DS) effectively suppresses HIF-1α expression and its downstream effectors, significantly reducing tumor cell migration and invasion in both in vitro and in vivo models (207). In PCa, HIF-1α-induced EMT relies on Wnt/β-catenin signaling, as β-catenin inhibition reverses EMT traits and reduces invasiveness, underscoring Wnt signaling as a key downstream effector of HIF-1α (208). In CRC, HIF-1α binds directly to hypoxia response elements (HREs) within the ZEB1 promoter, thereby driving ZEB1 upregulation and promoting EMT, migration, and invasion (209).
The strong correlation between HIF-1α and ZEB1 expression in clinical CRC specimens, together with their association with mesenchymal markers and inverse relationship with E-cadherin, further supports this axis as a driver of metastasis. These findings position HIF-1α as a central regulator of hypoxia-induced EMT, acting through interconnected pathways including NF-κB, TGF-β, Wnt/β-catenin, and ZEB1, thereby presenting multiple therapeutic targets to hinder tumor progression and metastasis.
HIF-1α has also emerged as a key mediator of EMT, immune evasion, angiogenesis, and metastasis across several cancers, including ovarian, NSCLC, and PCa. In ovarian cancer, ginsenoside 20(S)-Rg3 effectively suppresses hypoxia-induced EMT in vitro and in vivo by promoting ubiquitin-dependent degradation of HIF-1α. This suppression leads to reduced Snail expression, restoration of E-cadherin, and decreased vimentin levels, signifying EMT reversal (210). In NSCLC, inhibition of HIF-1α with PX-478 enhances the efficacy of anti-PD-1 immunotherapy by promoting T-cell-mediated apoptosis, increasing tumor-infiltrating lymphocytes (TILs), and reversing the immunosuppressive EMT phenotype by disrupting the HIF-1α/LOXL2 signaling axis (211).
Additionally, elevated HIF-1α expression is linked to reduced tumor-infiltrating lymphocytes (TILs) and poorer patient outcomes, supporting the rationale for combining HIF-1α inhibitors with immune checkpoint blockade in clinical settings. Furthermore, in NSCLC, endogenous hydrogen sulfide (H₂S), produced by the overexpressed enzymes CBS, CSE, and MPST, promotes EMT, migration, invasion, and angiogenesis by activating HIF-1α and upregulating VEGF. In contrast, pharmacological inhibition of these H₂S-generating enzymes suppresses tumor growth and angiogenesis in vivo (212). In PCa, CAFs promote EMT and enhance the metastatic potential of CSCs by upregulating HIF-1α and β-catenin signaling, thereby driving a highly aggressive mesenchymal phenotype (213). Notably, silencing β-catenin abolishes this effect, underscoring the critical role of the HIF-1α/β-catenin axis in CSC migration. Collectively, these findings emphasize the multifaceted role of HIF-1α as a central regulator of EMT, immune resistance, and metastatic progression, establishing it as a promising therapeutic target across multiple cancer types. Leucine-rich alpha-2-glycoprotein 1 (LRG1), previously implicated in several malignancies, has recently been identified as a key driver in CRC progression.
Analyses of CRC specimens revealed that LRG1 is significantly overexpressed and correlates with aggressive tumor features. Functional studies using HCT116 and SW480 cell lines demonstrated that LRG1 promotes migration and invasion by inducing EMT, characterized by reduced epithelial markers, such as E-cadherin and VDR, and increased mesenchymal markers, including N-cadherin, α-SMA, vimentin, and Twist1. In addition, LRG1 elevated VEGF-A production, thereby enhancing tumor angiogenesis, as confirmed through endothelial cell migration, tube formation, and aortic ring assays using conditioned media. Mechanistically, LRG1 was shown to upregulate HIF-1α in a dose- and time-dependent manner, thereby increasing VEGF-A expression and EMT-related changes. Collectively, these findings demonstrate that LRG1 drives CRC aggressiveness by activating HIF-1α signaling, thereby promoting EMT and angiogenesis, highlighting its potential as a therapeutic target for CRC management (214).
The poor prognosis of HCC is primarily driven by its highly invasive and metastatic potential, though the molecular mechanisms underlying these processes remain incompletely understood. Recent evidence shows that hypoxia accelerates HCC progression by inducing EMT through activation of HIF-1α. Elevated HIF-1α expression has been observed in HCC tissues and is strongly correlated with unfavorable clinical outcomes. Its overexpression is associated with increased mesenchymal markers such as N-cadherin and vimentin, reduced E-cadherin, and elevated SNAI1 levels.
Experimental studies have demonstrated that hypoxic conditions or CoCl₂ treatment promote EMT and significantly enhance the migratory and invasive capacities of HCC cells. Conversely, shRNA-mediated silencing of HIF-1α effectively reversed CoCl₂-induced EMT, reducing cell migration and invasion. Luciferase reporter assays further revealed that HIF-1α directly regulates SNAI1 transcription through two hypoxia response elements (HREs) within its promoter. Collectively, these findings demonstrate that hypoxia-induced stabilization of HIF-1α drives EMT by transcriptionally upregulating SNAI1, underscoring HIF-1α as a promising therapeutic target in HCC (215).
HCC continues to rise in both incidence and mortality, with tumor progression strongly influenced by the hypoxic microenvironment, a hallmark of solid tumors. Hypoxia drives epithelial-mesenchymal transition (EMT), vasculogenic mimicry (VM), and metastasis, all of which contribute to poor clinical outcomes. HIF-1α functions as a central transcriptional regulator under low-oxygen conditions and has been shown to upregulate lysyl oxidase-like 2 (LOXL2), an enzyme responsible for ECM remodeling through collagen and elastin cross-linking. In HCC tissues, elevated levels of HIF-1α and LOXL2 are positively correlated, indicating a poor prognosis and VM formation.
Functional studies demonstrated that HIF-1α enhances EMT, cell motility, invasion, and VM by increasing LOXL2 expression; conversely, silencing HIF-1α suppresses LOXL2 and inhibits these malignant features. Rescue experiments confirmed that LOXL2 is both necessary and sufficient for mediating HIF-1α’s effects on EMT and VM. Transcriptome microarray analyses further supported the role of LOXL2 in hypoxia-induced HCC transformation. Collectively, these findings highlight the HIF-1α/LOXL2 axis as a key driver of HCC progression, suggesting that targeting LOXL2 could represent a promising therapeutic strategy to restrain tumor growth and metastasis (216).
Tumor progression is profoundly influenced by the TME, particularly through hypoxia and inflammation, which converge to activate HIF-1α. HIF-1α is frequently overexpressed in cancers, where it drives hypoxia-responsive gene expression to support tumor survival and contributes to genomic instability by suppressing DNA repair. Chronic hypoxia induces a sustained EMT via HIF-1α-dependent upregulation of ZEB2, while transient hypoxia triggers a reversible EMT through Twist1. Cells undergoing these transitions acquire heightened tumorigenicity, necrosis, invasiveness, and metastatic potential. In parallel, interferon (IFN), a key inflammatory cytokine, promotes functional HIF-1α expression under normoxic conditions through the PI3K/AKT/mTOR pathway, thereby fostering EMT, resistance to apoptosis, vasculogenic mimicry, and the development of aggressive tumor phenotypes. Targeting HIF-1α or its downstream signaling pathways markedly reduces these malignant traits in vitro and in vivo. Collectively, these findings reveal a critical interplay between hypoxic and inflammatory signals in the TME, positioning HIF-1α as a central therapeutic target to block EMT, metastasis, and therapy resistance (217,218).
4.9. Nrf2 signaling pathway
EMT is a key driver of cancer metastasis, often characterized by a hybrid epithelial/mesenchymal (E/M) phenotype that enhances tumor aggressiveness and collective migration. The transcription factor NRF2 has been identified as a critical stabilizer of this hybrid state, as it simultaneously regulates both epithelial and mesenchymal markers, thereby contributing to metastasis and a poor prognosis. Computational and experimental studies demonstrate that NRF2 knockdown disrupts the hybrid phenotype and impairs collective migration, whereas its overexpression reinforces it. Moreover, cancer cells activate M2-like tumor-associated macrophages (TAMs) through NRF2 signaling induced by lactate-driven ROS. In turn, these macrophages promote EMT in cancer cells by reactivating NRF2 via VEGF signaling. Collectively, these findings establish NRF2 as a central regulator of hybrid E/M states and macrophage-cancer cell interactions, highlighting its potential as a therapeutic target (219,220).
EMT is a biological process in which epithelial cells acquire mesenchymal properties, playing a pivotal role in cancer progression. Notch signaling is a key mediator of TGF-β1-induced EMT, primarily through the direct transcriptional activation of Snai1. However, the mechanism by which TGF-β1 activates Notch signaling remains insufficiently defined. Emerging evidence suggests that the ROS-Nrf2 pathway plays a crucial role in regulating this process. TGF-β1 increases ROS production, which in turn activates Nrf2. Inhibition of Nrf2 activity, either through ROS scavenging with N-acetylcysteine or Nrf2-targeted small interfering RNA (siRNA), reduces Notch signaling and attenuates EMT progression. Furthermore, TGF-β1 upregulates Notch4 transcription by inducing Nrf2-dependent promoter activation. Collectively, these findings identify the ROS-Nrf2 axis as a central mediator of TGF-β1-driven EMT, activating Notch signaling (221). Figure 4 illustrates the complex signaling networks that regulate EMT in cancer.

Figure 4: The complicated networks of factors regulating EMT in cancer. Multiple pathways, including PI3K/Akt/mTOR, STAT3, Wnt/β-catenin, TGF-β1, Sonic Hedgehog (Shh), Notch1, TNF-α, and hypoxia-inducible factor 1α (HIF-1α), converge to activate EMT programs. These signals stimulate EMT-inducing transcription factors such as Snail, Slug, Twist, and ZEB family proteins, thereby promoting loss of epithelial characteristics, acquisition of mesenchymal traits, and enhanced cellular plasticity. Through these mechanisms, EMT contributes to tumor invasion, metastasis, immune evasion, and therapy resistance. The figure highlights how interconnected regulatory cascades orchestrate EMT, underscoring its central role in cancer progression and its potential as a therapeutic target.
5. Non-coding RNA-driven regulation of EMT in cancer
5.1. miRNA
In ovarian cancer, the transcription factor Snail is a well-established driver of EMT, a key step in metastasis. However, the regulation of Snail expression by specific miRNAs remains insufficiently understood. Computational target prediction identified six candidate miRNAs targeting the 3′-untranslated region (3′-UTR) of Snail, with miR-137 and miR-34a emerging as the strongest candidates. Experimental validation using quantitative real-time PCR, Western blotting, and 3′-UTR luciferase reporter assays confirmed their direct binding to Snail mRNA. Functional studies showed that overexpression of miR-137 or miR-34a suppressed Snail at both transcript and protein levels, thereby inhibiting EMT, invasion, and sphere formation in ovarian cancer cells. Conversely, inhibition of these miRNAs via antisense oligonucleotides enhanced EMT and invasive behavior. Significantly, ectopic Snail expression reversed the suppressive effects of miR-137 and miR-34a, confirming their regulatory role. Clinical analyses further revealed that miR-137 and miR-34a are significantly downregulated in ovarian cancer tissues compared with adjacent normal tissues, and their reduced expression correlates with poor patient survival. Collectively, these findings establish miR-137 and miR-34a as tumor-suppressive regulators of EMT and metastasis in OC through direct targeting of Snail (222).
MiRNAs are small, non-coding RNAs that can act as either oncogenes or tumor suppressors across various human cancers. Increasing evidence indicates that miRNA dysregulation plays a role in the initiation and progression of non-small cell lung cancer (NSCLC). Among these, miR-132 is significantly downregulated in both NSCLC cell lines and clinical tumor samples. Restoring miR-132 expression markedly suppresses the migration and invasion of lung cancer cells in vitro, supporting its potential tumor-suppressor role. Mechanistic studies revealed that miR-132 directly targets ZEB2, a transcription factor closely linked to EMT, by binding to its 3′ untranslated region (3′ UTR). miR-132 negatively regulates ZEB2 at both transcript and protein levels, leading to increased E-cadherin and reduced vimentin expression, hallmarks of EMT reversal. Functional assays demonstrated that modulating ZEB2 expression mimics the effects of miR-132, with ZEB2 overexpression enhancing, and ZEB2 silencing suppressing, cell motility and invasion. Furthermore, ZEB2 knockdown offsets the pro-invasive effects triggered by miR-132 inhibition. Collectively, these findings indicate that miR-132 restrains NSCLC cell migration and invasion by directly targeting ZEB2 and regulating EMT, suggesting its promise as a therapeutic target in lung cancer management (223).
EMT plays a central role in cancer progression, metastasis, and therapy resistance, with growing evidence highlighting the regulatory influence of specific miRNAs in this process across multiple tumor types. In chemotherapy-resistant TSCC, long-term exposure to cisplatin led to the establishment of stable cell lines that exhibited mesenchymal traits, increased motility, and reduced expression of miR-200b and miR-15b (224). Restoring these miRNAs reversed EMT, suppressed invasion, and increased chemosensitivity by directly targeting BMI1, whereas their inhibition induced EMT and promoted drug resistance. In vivo, enforced expression of miR-200b or miR-15b effectively reduced metastasis, whereas their downregulation correlated with therapy resistance and poorer survival outcomes in clinical samples. Similarly, miR-655 has been identified as an EMT suppressor in pancreatic and esophageal cancers, where it upregulates E-cadherin and impedes migration by targeting ZEB1 and TGFBR2 (225). In PCa, SLUG promotes EMT and tumor progression by repressing miR-1 and miR-200 in a feedback loop that amplifies EMT (226); restoring these miRNAs effectively hinders both EMT and tumorigenesis. In breast cancer, miR-506 has been identified as a key regulator associated with improved survival outcomes, as it suppresses mesenchymal markers, including Vimentin and Snai2, and inhibits TGF-β-induced EMT and invasive behavior (227). These findings highlight the critical role of EMT-regulating miRNAs, particularly miR-200b, miR-15b, miR-655, miR-1, miR-200, and miR-506, in controlling metastasis, therapy resistance, and cancer progression by regulating EMT-associated transcription factors and signaling pathways, thereby establishing them as promising therapeutic targets across multiple malignancies.
5.2. LncRNA
Gastric cancer is a highly heterogeneous malignancy and a major global health burden, with the roles of lncRNAs in its various subtypes still poorly understood. Among these, the microsatellite-stable (MSS)/EMT subtype is the most aggressive and is linked to poor clinical outcomes. Through integrated network analysis, MIR200CHG has been identified as a key long non-coding RNA (lncRNA) regulator of the epithelial-mesenchymal transition in this subtype. Its expression is often suppressed by promoter hypermethylation, a marker of an unfavorable prognosis. Functionally, MIR200CHG reduces mesenchymal features in gastric cancer cells in vitro and inhibits metastasis in vivo. Mechanistically, MIR200CHG enhances the production of its intronic miRNAs, miR-200c and miR-141, and directly interacts with miR-200c to protect it from degradation via target-directed miRNA degradation (TDMD). These findings reveal a subtype-specific long non-coding RNA (lncRNA) regulatory network, providing important biological insights into the progression of MSS/EMT gastric cancer (228).
Metastasis is a complex, multistep process in which tumor cells spread from their primary site to establish secondary tumors in distant organs. This progression is driven by the phenotypic plasticity of cancer cells, allowing them to transition between epithelial and mesenchymal states through transcriptionally regulated processes such as EMT and its reverse, MET. Using a mouse model of spontaneous metastatic breast cancer, researchers examined the molecular regulators of metastatic competence within a heterogeneous primary tumor and the mechanisms by which cells dynamically adjust epithelial-mesenchymal plasticity during metastasis.
Cells isolated from primary mammary tumors, circulating blood, and lung metastases in TA2 mice revealed that the long noncoding RNA (lncRNA) H19 promotes both EMT and MET by acting as a molecular sponge for miR-200b/c and let-7b. This interaction regulates the expression of GIT2 and CYTH3, key modulators of the ARF GTPase family, which are critical for cell migration during EMT and metastatic dissemination. Silencing H19 or disrupting its associated pathway significantly reduced metastasis in syngeneic mouse models.
Furthermore, elevated levels of H19, GIT2, and CYTH3 in patient tumor samples highlight H19’s potential as both a biomarker of metastatic breast cancer and a therapeutic target for limiting metastatic progression (229). The TGF-β/Smad signaling pathway plays a central role in driving EMT during cancer progression. Increasing evidence indicates that lncRNAs are key regulators of cancer cell behavior, including EMT. A recently identified long non-coding RNA located near Smad3, termed Smad3-associated lncRNA (SMASR), functions as a negative regulator of EMT in lung cancer. TGF-β suppresses SMASR expression through the Smad2/3 signaling pathway. Loss of SMASR promotes EMT, thereby enhancing the migratory and invasive capacities of lung cancer cells. Mechanistically, SMASR knockdown increases Smad2/3 phosphorylation, reflecting heightened TGF-β pathway activity. Further analysis revealed that SMASR directly interacts with Smad2/3 and downregulates TGFBR1, the type I TGF-β receptor that phosphorylates Smad2/3, thereby attenuating the TGF-β/Smad cascade. Clinically, lung cancer tissues display reduced SMASR expression. Collectively, these findings identify SMASR as a critical inhibitor of EMT, acting through a negative feedback loop within the TGF-β/Smad signaling pathway (230).
5.3. CircRNA
Bladder cancer is a prevalent urological malignancy marked by aggressive invasion, frequent metastasis, recurrence, and resistance to therapy. Recent evidence suggests that circRNA-protein complexes play a critical role in tumor progression; however, their precise contribution to bladder cancer (BCa) metastasis and chemoresistance remains insufficiently defined. Among these, circPTK2 has been identified as an upregulated circular RNA that regulates SETDB1 expression, as revealed by RNA sequencing. CircRNA pulldown assays and RNA-binding protein immunoprecipitation further identified PABPC1 as a primary binding partner of circPTK2. Mechanistically, circPTK2 interacts with PABPC1, enhancing PABPC1's ability to stabilize SETDB1 mRNA and thereby promoting SETDB1 expression. This upregulation drives SETDB1-mediated EMT, fueling tumor progression. Functionally, the circPTK2-PABPC1-SETDB1 axis significantly enhances cell migration, invasion, and gemcitabine resistance in vitro and promotes lymph node metastasis in vivo. These findings reveal a previously unrecognized role of this signaling pathway in EMT-driven metastasis and chemoresistance in bladder cancer (231).
Lung adenocarcinoma (LUAD) is a leading cause of cancer incidence and mortality worldwide, yet its underlying molecular mechanisms are still not fully understood. Circular RNAs (circRNAs) are recognized as essential regulators in cancer biology, although their specific roles in LUAD remain to be elucidated. Microarray analysis identified circ-HMGA2 (hsa_circ_0027446) as a novel circRNA significantly upregulated in LUAD, a finding further confirmed in 36 paired LUAD and adjacent normal tissue samples.
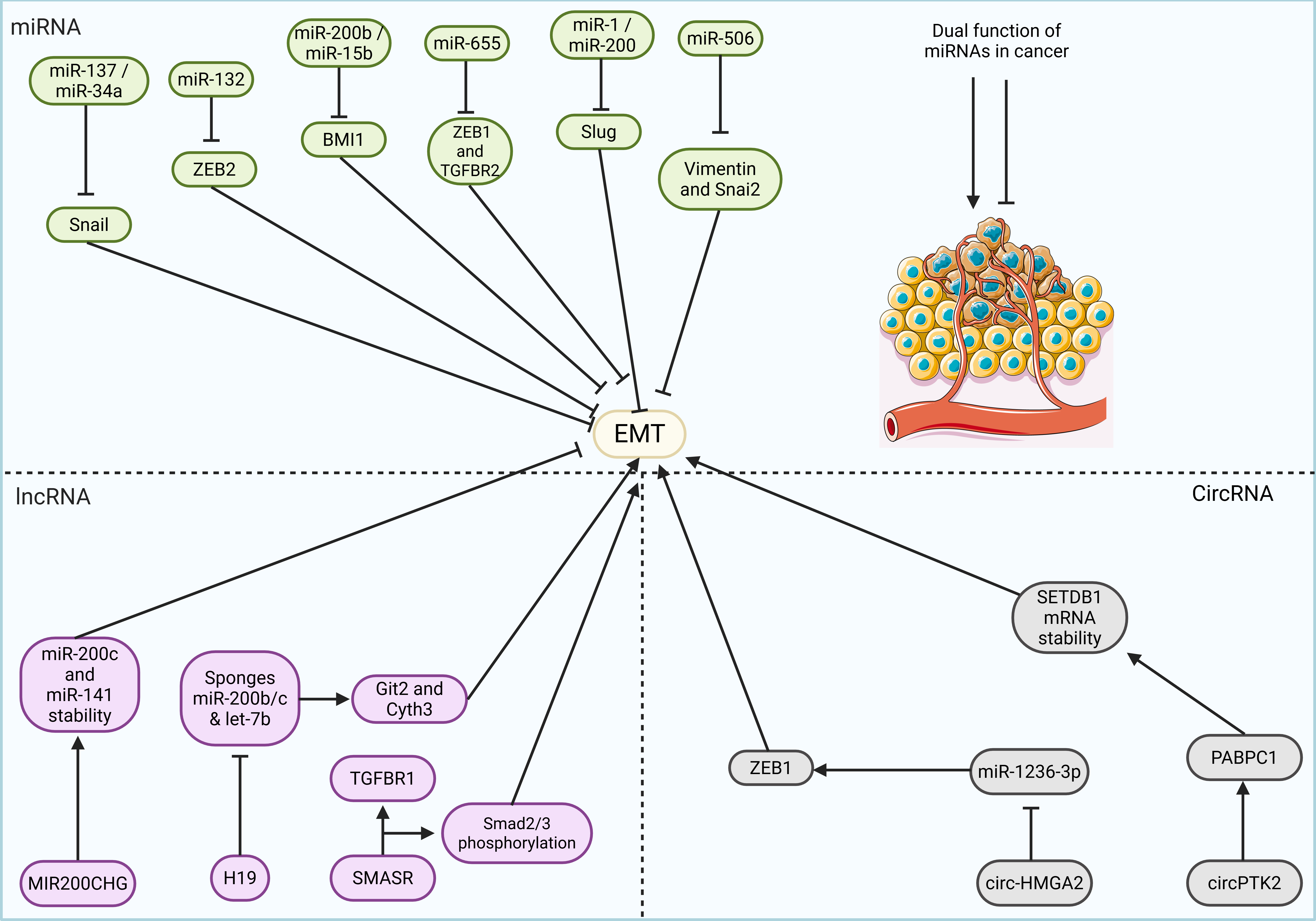
Figure 5: Schematic illustration of non-coding RNA-mediated regulation of EMT in cancer. Multiple miRNAs, lncRNAs, and circRNAs modulate EMT by directly targeting EMT-inducing transcription factors or signaling molecules. Tumor-suppressive miRNAs such as miR-137, miR-34a, miR-132, miR-200b, miR-15b, miR-655, miR-1, miR-200, and miR-506 inhibit key EMT drivers (Snail, ZEB2, BMI1, ZEB1, TGFBR2, SLUG, vimentin, Snai2) to restore epithelial markers and suppress invasion, metastasis, and drug resistance. LncRNAs exhibit both EMT-suppressive (MIR200CHG, SMASR) and EMT-promoting (H19) functions by modulating miRNA stability, sponging tumor-suppressive miRNAs, or regulating TGF-β/Smad signaling. CircRNAs such as circPTK2 and circ-HMGA2 promote EMT and metastasis by stabilizing mRNA transcripts (SETDB1) or sponging miRNAs (miR-1236-3p) that target EMT-related transcription factors (ZEB1). These non-coding RNA networks orchestrate EMT plasticity, influencing cancer progression, therapeutic resistance, and patient prognosis.
Functionally, circ-HMGA2 was shown to promote LUAD cell metastasis both in vitro and in vivo. Luciferase reporter assays and fluorescence in situ hybridization (FISH) demonstrated that circ-HMGA2 directly binds to miR-1236-3p, which generally targets ZEB1, a transcription factor known to drive EMT. MiR-1236-3p was downregulated in LUAD and exhibited tumor-suppressive effects by reducing metastasis and counteracting the activity of circ-HMGA2. Further analysis using PCR and Western blotting revealed that circ-HMGA2 upregulates ZEB1 expression and promotes EMT, whereas miR-1236-3p mitigates this effect by suppressing the EMT-inducing function of circ-HMGA2 (232). Figure 5 demonstrates the function of non-coding RNAs in the regulation of EMT in cancer.
6. EMT and immune evasion
The TME defined by hypoxia, acidity, inflammation, and immunosuppression plays a central role in cancer progression by driving EMT. EMT not only enhances tumor cell plasticity but also reprograms cancer cells to evade immune surveillance.
Tumor-associated macrophages (TAMs), which predominantly acquire an M2-like pro-tumor phenotype, promote EMT, matrix remodeling, and angiogenesis through multiple signaling routes, including TGF-β, COX-2/STAT, EGFR/ERK1/2, Smad/Snail, AKT/mTOR, CCL-18, and HIF-1α. Under hypoxic conditions, HIF-1α stimulates tumor-derived CCL-20 production, which, in turn, elevates indoleamine 2,3-dioxygenase (IDO) expression in macrophages, thereby suppressing the activity of CD4+ and CD8+ T cells. Notably, mTORC1/2 inhibition reduces EMT, TAM recruitment, and PD-L1 expression.
Myeloid-derived suppressor cells (MDSCs) also impair the function of T cells, natural killer cells (NK cells), and dendritic cells (DCs). Their infiltration is largely driven by CXCL2 and COX-2. Snail, a key EMT transcription factor (EMT-TF), activates CXCL2 via the NF-κB pathway, thereby attracting MDSCs, which further reinforce EMT through β-catenin/TCF4 and COX-2 signaling, thereby promoting metastasis.
Beyond this, EMT-TFs directly shape immune evasion. Snail expands Tregs, fosters resistance to immunotherapy, and recruits Tregs, TAMs, and MDSCs through CCL2, while generating immunosuppressive DCs. Twist, another EMT-TF, is upregulated by PD-L1 and MFG-E8, promoting Treg infiltration, inhibiting CD8+ and NK cell activity, and modulating TNFα/NF-κB via type I interferons. Similarly, ZEB1 and ZEB2 influence DC activation, NK maturation, and cytotoxic T lymphocyte (CTL) function. Through the ZEB1/miR-200 axis, they mediate the PD-L1-driven suppression of CD8+ T cells.
Cancer cell plasticity further enhances immune evasion. Mesenchymal-like states confer resistance to CTL- and NK-mediated lysis, which is aided by autophagy and by pathways such as brachyury and estrogen receptor signaling. However, context-specific exceptions exist during metastatic stages; EMT can increase NK-activating ligands or CADM1, temporarily improving NK surveillance. Importantly, TGF-β blockade can restore NK sensitivity in mesenchymal subclones.
Clinically, EMT is associated with elevated PD-L1 and reduced E-cadherin expression, correlating with poor survival in PD-L1+/EMT+ tumors. This suggests EMT status may serve as a co-biomarker alongside PD-L1 to predict immunotherapy outcomes, given that PD-L1 expression alone is insufficient as a response marker. Snail-positive, mesenchymal-like tumors frequently inhibit antigen presentation by downregulating MHC class I and β2-microglobulin, thereby reducing CD8+ T-cell recognition. In melanoma, Snail1 inversely correlates with HLA-I transcription, as indicated by ChIP studies, which show Snail recruitment to MHC-I promoters. In PC, TGF-β is required for Snail-mediated HLA-I suppression, as Snail (either ectopic or induced by TGF-β) downregulates NF-κB, a key regulator of HLA-I.
Other EMT inducers, such as AXL, also contribute to immune evasion. AXL overexpression correlates with reduced MHC-I in NSCLC, melanoma, and breast cancer, whereas AXL silencing restores epithelial traits and enhances tumor clearance by reinstating MHC-I expression. EMT-linked stemness also correlates with MHC-I downregulation, thereby fostering an immunologically “cold” tumor environment that is resistant to CTL attack.
Therapeutically, inhibiting EMT in metastatic breast cancer models restores MHC-I and improves responsiveness to PD-L1 blockade. These findings emphasize that EMT-driven reprogramming suppresses antigen presentation, weakens CTL surveillance, and fosters immune escape, thereby underscoring EMT as a critical target for enhancing the efficacy of immunotherapy (233).
An increasing body of evidence demonstrates the coexistence of EMT and immune regulation; however, a comprehensive systems-level analysis of their interplay and its implications for tumor progression and clinical outcomes across cancer types remains limited. Large-scale multi-omics studies encompassing 17 solid tumors have revealed a conserved, dynamic interaction between EMT and immune evasion, which is strongly linked to patient prognosis and response to immunotherapy. Integrative genomic and immunogenomic analyses further reveal that factors such as immune infiltrate composition, nonsynonymous mutation burden, chromosomal instability, and oncogenic alterations shape the balance between EMT and immune escape. To quantify this relationship, researchers developed the EMT-CYT Index (ECI), a scoring model that demonstrated superior predictive power for survival and treatment outcomes across multiple malignancies. These findings position EMT-immunity interactions as a pan-cancer paradigm for understanding the molecular drivers of progression and for guiding the development of broadly applicable immunotherapy strategies.
Supporting this, CTCs from patients with gastric cancer exhibit distinct EMT phenotypes. Specifically, mesenchymal-like CTCs display significantly reduced expression of ULBP1, a critical ligand required for natural killer (NK) cell recognition, compared with epithelial-like CTCs. Analysis of 41 patient samples using Cyttel-CTC and im-FISH, in conjunction with gastric cancer cell line studies, confirmed that TGF-β-induced EMT similarly suppresses ULBP1 expression. This suggests that EMT enables CTCs to escape NK cell-mediated immune surveillance. Collectively, these findings underscore EMT as a central mechanism of immune evasion in CTCs, highlighting potential therapeutic opportunities to restore NK cell recognition by reversing EMT-driven suppression of ULBP1 (234).
EMT plays a central role in driving PCa invasion and dissemination, yet the precise mechanisms underlying EMT in clinical settings remain insufficiently defined. In this context, N-cadherin expression is considered a key indicator of EMT. To clarify its role, laser capture microdissection was performed on paired N-cadherin-positive and negative tumor regions from prostate cancer (PCa) patients (n = 8), followed by RNA sequencing. The analysis revealed a strong correlation between N-cadherin expression and an immune-regulatory signature, most notably characterized by a marked upregulation of indoleamine 2,3-dioxygenase (IDO1; log2-fold change = 5.1; P = 2.98E-04). Immunofluorescent labeling further confirmed the elevated expression of IDO1 protein and its metabolite, kynurenine, in predominantly N-cadherin-positive regions. These same areas exhibited reduced intraepithelial cytotoxic CD8+ T-cell infiltration, accompanied by an enrichment of immunosuppressive CD4+/FOXP3+ regulatory T cells. Collectively, these findings demonstrate that EMT in clinical PCa is associated with heightened IDO1 expression and the expansion of regulatory T cells, suggesting that EMT-driven changes promote immune evasion and protect tumor growth from effective immune surveillance (235).
7. Natural compounds and EMT in cancer
Tobacco smoke (TS) is the primary risk factor for bladder cancer, with EMT recognized as a key process in TS-associated tumorigenesis. CSCs also play a central role in tumor progression, particularly under TS exposure. However, the molecular mechanisms linking TS to EMT induction and the development of CSC-like traits remain incompletely defined. The Wnt/β-catenin signaling pathway is critical for regulating EMT and maintaining CSC properties. Prolonged TS exposure has been shown to induce EMT-related changes and significantly increase the expression of CSC markers. Evidence indicates that TS enhances Wnt/β-catenin signaling, thereby driving EMT and stemness, whereas inhibiting this pathway suppresses these effects. Notably, curcumin treatment effectively blocks TS-induced Wnt/β-catenin activation, reversing EMT features and CSC acquisition. Collectively, these findings emphasize the regulatory role of Wnt/β-catenin in TS-mediated bladder cancer progression and highlight the potential of curcumin as a chemopreventive strategy (236).
Epigallocatechin-3-gallate (EGCG), the primary polyphenol in green tea, exhibits potent anticancer activity across multiple malignancies, primarily by targeting CSCs and EMT, two critical processes in tumor initiation, progression, invasion, metastasis, and therapeutic resistance. Studies in nasopharyngeal, pancreatic, prostate, and oral squamous cell carcinomas consistently show that EGCG suppresses CSC self-renewal, sphere formation, invasion, and migration, while inducing apoptosis by downregulating anti-apoptotic proteins such as Bcl-2, survivin, and XIAP, and activating caspases. EGCG also blocks EMT by reducing the expression of vimentin, Snail, Slug, β-catenin, and TCF/LEF signaling, while inhibiting MMP-2, MMP-9, urokinase-type plasminogen activator, p-FAK, and p-Src. In PC, EGCG prevents the cadherin switch and inhibits Akt signaling independently of oxidative stress, and it shows synergistic effects when combined with gemcitabine. In PCa, co-treatment with EGCG and quercetin markedly enhances CSC inhibition and induces apoptosis. Collectively, these findings highlight EGCG’s ability to eliminate CSC populations, suppress EMT, and reduce migration and invasion, reinforcing its promise as both a dietary chemopreventive and therapeutic agent across diverse cancers (237-240).
Kaempferol, a naturally occurring dietary flavonoid with recognized chemopreventive and anticancer properties, exhibits potent antimetastatic activity by inhibiting EMT and cancer cell migration in multiple contexts. In MCF-7 breast cancer cells, kaempferol counteracted the EMT-inducing and pro-migratory effects of triclosan, a xenoestrogenic endocrine disruptor, as well as estrogen (E2), by restoring epithelial morphology, reducing migration and invasion, and suppressing EMT- and metastasis-associated markers through estrogen receptor antagonism. Similarly, in A549 non-small cell lung cancer cells, kaempferol inhibited TGF-β1-induced EMT and migration by preserving E-cadherin, reducing mesenchymal markers, and suppressing MMP-2 activity. Mechanistically, kaempferol blocked TGF-β1-induced phosphorylation of Smad3 at Thr179, a modification critical for EMT, by preventing Akt1 activation, while leaving Smad3 nuclear translocation and complex formation unaffected. Collectively, these findings provide strong molecular evidence for kaempferol’s role in suppressing EMT and metastasis in both hormone-dependent and TGF-β1-driven cancers (241,242).
Quercetin, a naturally occurring flavonoid, exhibits significant anticancer activity against multiple malignancies, primarily by inhibiting EMT, a key driver of cancer metastasis. In NSCLC, quercetin was shown to suppress cell migration, invasion, and bone metastasis by blocking Snail-driven EMT and Akt activation, while concurrently modulating maspin and ADAM9 expression. These effects contributed to prolonged survival in animal models, highlighting quercetin’s promise as a therapeutic candidate (243). Clinical evidence supports these findings, showing poorer outcomes in patients with elevated Snail/p-Akt or ADAM9 expression.
In CRC models, quercetin markedly suppressed TGF-β1-induced EMT and invasiveness in SW480 cells by downregulating Twist1, underscoring its potential to hinder EMT progression and metastatic capacity (244). In PCa, treatment with quercetin suppressed the oncogenic lncRNA MALAT1, inhibited tumor cell proliferation and EMT, promoted apoptosis, and blocked activation of the PI3K/Akt pathway in both in vitro and in vivo models (245). Overexpression of MALAT1 markedly conferred resistance to quercetin, highlighting its mechanistic role in modulating the drug’s anticancer activity. These results suggest that quercetin suppresses cancer progression and metastasis through multiple molecular mechanisms, including the inhibition of EMT, induction of apoptosis, and modulation of key signaling pathways, thereby reinforcing its promise as a multifaceted anticancer agent.
Resveratrol, a natural compound with established anticancer properties, has significant potential to suppress EMT, a key driver of cancer progression and metastasis. In ovarian cancer cells, Resveratrol was found to counteract Cisplatin-induced EMT by downregulating Snail expression via ERK signaling pathway inhibition, reversing EMT-associated morphological changes, and reducing cell migration, while inducing cell death through an apoptosis-independent mechanism (246). In MCF-7 breast cancer cells, Resveratrol markedly suppressed EGF-induced EMT by preventing morphological and motility changes and by downregulating key mesenchymal markers and transcription factors, such as Slug, Zeb1, and Zeb2 (247). Mechanistic studies revealed that both cisplatin- and EGF-induced EMT are regulated by ERK1/2 signaling, with Resveratrol’s ability to inhibit ERK activation a key factor in its anti-EMT effects. Collectively, these findings highlight the potential of Resveratrol as a valuable adjunct to chemotherapy, capable of mitigating EMT-driven metastasis across multiple cancer types.
Berberine alkaloids, a group of isoquinoline compounds, have demonstrated potent anticancer activity, particularly in inhibiting breast cancer progression. While berberine itself is known to suppress breast cancer growth, the mechanisms underlying the actions of its derivatives epiberberine, berberrubine, and dihydroberberine remain less well defined. Experimental assays, including MTT, colony formation, wound-healing, and Transwell invasion assays, demonstrated that these derivatives significantly reduced the proliferation, migration, and invasion of breast cancer cells. Apoptosis analysis (Hoechst and Annexin V-FITC/PI staining) and Western blotting revealed modulation of proteins linked to the Wnt/β-catenin pathway and EMT. These alkaloids inhibited proliferation by inducing cell-cycle arrest at the G2/M phase in MCF-7 cells and at both the G0/G1 and G2/M phases in MDA-MB-231 cells. Mechanistically, they downregulate β-catenin, upregulate GSK-3β, decrease N-cadherin, and increase E-cadherin, thereby suppressing metastasis and reversing EMT, though berberine itself did not fully induce EMT reversal. Additionally, these compounds induce apoptosis via both intrinsic and extrinsic pathways, underscoring their therapeutic potential in breast cancer treatment (248).
Natural compounds such as genistein, apigenin, and sulforaphane have demonstrated promising antimetastatic activity across various cancers by modulating EMT and its associated signaling pathways. In HCC cells, genistein suppressed migration and EMT in a dose-dependent manner by upregulating epithelial markers (E-cadherin, α-catenin) while downregulating mesenchymal markers (N-cadherin, vimentin) (249). This effect was partly mediated through inhibition of the NFAT1 pathway and was confirmed in both in vitro and in vivo models. In estrogen-responsive BG-1 ovarian cancer cells, genistein reversed estrogen-induced EMT and migration, induced by estrogenic compounds such as E2, BPA, and NP, by acting through estrogen receptors and TGF-β signaling pathways (250).
Apigenin, a flavonoid, significantly suppresses proliferation, migration, and stemness in TNBC by disrupting the YAP/TAZ-TEAD protein-protein interaction and downregulating downstream target genes such as CTGF and CYR61 (251). In PCa, apigenin exerts anti-metastatic effects by downregulating SPOCK1, a key regulator of EMT, and reducing mesenchymal marker expression, thereby inhibiting tumor progression and prolonging survival in animal models (252).
Sulforaphane, a bioactive compound abundant in cruciferous vegetables, exerts potent anti-cancer effects by suppressing the migration and invasion of NSCLC cells through downregulation of miR-616-5p, a miRNA associated with advanced-stage disease and metastasis (253). Sulforaphane suppresses miR-616-5p expression through histone modification, restores GSK3β levels, and inactivates the GSK3β/β-catenin pathway, thereby inhibiting EMT and reducing metastasis in vivo. Collectively, these findings highlight the therapeutic potential of naturally derived compounds as adjuvant strategies in cancer treatment, targeting EMT and metastasis-associated pathways.
8. Nanoparticles and EMT in cancer
EMT is widely recognized as a critical driver of breast cancer metastasis and chemoresistance. MCF-7/ADR cells exhibit more pronounced EMT characteristics than their parental MCF-7 cells, resulting in enhanced drug resistance through increased P-gp expression and decreased apoptotic sensitivity. Suppressing EMT in these cells decreases P-gp expression and apoptosis-related protein levels, thereby restoring the cytotoxic activity of doxorubicin (DOX). Although emodin (EMO) has shown promise in inhibiting EMT and overcoming drug resistance, its poor cellular uptake limits clinical application. To address this, emodin-loaded polymer-lipid nanoparticles (E-PLNs) significantly improve EMO internalization and effectively block EMT in MCF-7/ADR cells. This intervention resensitizes the cells to DOX, reducing resistance and enhancing treatment efficacy. Collectively, these findings suggest that E-PLNs enhance DOX responsiveness in breast cancer by inhibiting EMT. The combined use of E-PLNs and DOX may represent a promising therapeutic strategy to improve chemotherapy outcomes and delay or prevent the onset of resistance in breast cancer management (254).
TWIST, a transcription factor essential for embryonic development, is aberrantly activated in several cancers, where it serves as a master regulator of EMT and is strongly associated with angiogenesis, metastasis, CSC traits, and chemoresistance. In epithelial ovarian cancer, although most patients with metastatic disease initially respond to first-line therapies, the majority eventually relapse. These recurrences are typically characterized by renewed metastasis and acquired drug resistance, contributing to a dismal five-year survival rate of less than 20%. Recent research demonstrates that silencing TWIST using siRNA (siTWIST) delivered via an advanced nanoparticle system can effectively reverse chemoresistance in epithelial ovarian cancer models. Specifically, mesoporous silica nanoparticles conjugated to hyaluronic acid (MSN-HAs) were used to deliver siTWIST selectively into cancer cells, resulting in sustained suppression of TWIST expression in vitro. In vivo, mice treated with the combination of siTWIST-MSN-HA and cisplatin exhibited precise tumor targeting and a significant reduction in tumor burden. Collectively, these findings highlight the therapeutic potential of this strategy for overcoming key clinical obstacles, including selective delivery, metastasis, and chemoresistance in ovarian and other TWIST-driven cancers (255).
Natural bioactive compounds are increasingly recognized as promising antitumor and antimicrobial agents, valued for their strong therapeutic potential and relatively low systemic toxicity. However, their clinical application is hindered by limitations, including poor bioavailability and inadequate tumor targeting. To address these challenges, the development of safe, biocompatible nanoparticle-based delivery systems is critical for efficient drug transport to tumor tissues. In this study, amine-functionalized zinc oxide nanoparticles were engineered to encapsulate luteolin. These modified nanoparticles demonstrated enhanced synergistic effects, exhibiting both potent anticancer and antibacterial activity. Importantly, they selectively targeted cancer cells by inducing significant oxidative stress while exerting minimal cytotoxicity on normal cells. Mechanistic investigations revealed that their antitumor activity was mediated through the induction of autophagy and suppression of EMT (256).
The development of effective delivery systems for siRNA remains a significant challenge in cancer therapy. Nanoparticle-based approaches have emerged as a promising strategy to overcome this limitation. In this context, poly(lactide-co-glycolide) nanoparticles were utilized to encapsulate DCAMKL-1-specific siRNA (NP-siDCAMKL-1) for targeted delivery in CRC models. Treatment with NP-siDCAMKL-1 in HCT116 tumor xenografts significantly inhibited tumor growth, accompanied by downregulation of the proto-oncogenes c-Myc and Notch-1 via miRNA-dependent mechanisms involving let-7a and miR-144. In vitro assays further confirmed reduced luciferase activity at let-7a and miR-144 binding sites, supporting the specificity of these interactions.
Additionally, NP-siDCAMKL-1 increased the expression of the EMT inhibitor miR-200a while reducing the expression of EMT-associated transcription factors, such as ZEB1, ZEB2, Snail, and Slug. The administration of DAPT, a γ-secretase inhibitor, further enhanced tumor suppression by downregulating Notch-1 in a miR-144-dependent manner. Collectively, these findings highlight the therapeutic potential of nanoparticle-mediated siRNA delivery to modulate oncogenic signaling and endogenous miRNAs, offering a novel strategy for the treatment of colorectal and potentially other cancers (257).
Acquisition of the EMT phenotype in cancers is associated with reduced chemotherapy effectiveness and poorer prognosis. A previous study investigated the application of clinically available therapeutic ultrasound techniques to enhance the delivery of nanotherapeutics in both epithelial and EMT tumors, using positron emission tomography (PET), labeled nanoparticles for tracking. Epithelial tumors displayed dense vascularization and intact cell-cell junctions, whereas EMT tumors exhibited an elongated, irregular morphology, weakened intercellular adhesion, and reduced expression of E-cadherin and cytokeratins 8, 18, and 19. In the absence of ultrasound, in vivo accumulation of liposomal nanoparticles was approximately 1.5-fold higher in epithelial tumors compared with EMT tumors. Ultrasound treatment enhanced nanoparticle uptake and tumor permeability in both tumor types, with the degree of enhancement varying according to the applied thermal and mechanical indices. Notably, higher thermal ultrasound doses led to greater nanoparticle accumulation in EMT tumors (258).
Metastatic breast cancer remains the second leading cause of cancer-related mortality in women, with TNBC representing an especially aggressive subtype lacking clearly defined molecular targets for effective therapy. Conventional chemotherapy often fails due to high rates of recurrence, metastasis, and drug resistance. Given the established link between β3 integrin (ITGB3), EMT, and metastasis, β3 integrin was therapeutically targeted with lipid-based ECO nanoparticles that delivered ITGB3-specific siRNA (ECO/siβ3). Treatment of TNBC cells with ECO/siβ3 effectively suppressed ITGB3 expression, blocked TGFβ-induced EMT and invasion, restored TGFβ-mediated cytostasis, and reduced growth in three-dimensional organoid cultures. To enhance delivery efficiency, ECO/siβ3 nanoparticles were further functionalized with an RGD peptide via a PEG spacer, which increased siRNA uptake in post-EMT cells. In vivo, intravenous administration of RGD-conjugated ECO/siβ3 nanoparticles significantly reduced primary tumor burden and, critically, prevented metastasis. In a 16-week study that included primary tumor resection at week 9 and treatment discontinuation during the final 4 weeks, mice bearing orthotopic, TGFβ-prestimulated MDA-MB-231 tumors that received RGD-targeted ECO/siβ3 therapy showed no evidence of metastasis or recurrence, in stark contrast to untreated controls (259).
Discussion
EMT is a cornerstone of cancer biology, central to the mechanisms of tumor progression, invasion, metastasis, and therapy resistance. This cellular reprogramming process endows cancer cells with heightened plasticity, allowing them to detach from primary tumors, survive in hostile microenvironments, and colonize distant sites. At the core of EMT are transcription factors such as Snail, Slug, Twist, and Zeb, which coordinate the suppression of epithelial markers (e.g., E-cadherin) and the induction of mesenchymal markers (e.g., vimentin, N-cadherin). These molecular shifts are tightly regulated by interconnected signaling pathways, including TGF-β, Wnt/β-catenin, Notch, PI3K/Akt, and STAT3. The TME shaped by hypoxia, inflammatory mediators, and non-coding RNAs further modulates EMT activation and plasticity.
Beyond metastasis, EMT profoundly influences therapeutic outcomes and tumor recurrence. It is closely associated with drug resistance through mechanisms such as enhanced survival signaling, reduced apoptosis, altered drug transporter activity, and enrichment of CSC populations. Collectively, these properties position EMT as a pivotal driver of cancer aggressiveness and a critical target for therapeutic intervention.
The reversible and context-dependent nature of EMT, including the presence of hybrid epithelial/mesenchymal phenotypes, complicates therapeutic interventions and poses a significant challenge in clinical oncology. Partial EMT contributes to intratumoral heterogeneity and immune evasion, thereby enhancing tumor adaptability and resistance to conventional therapies. Thus, EMT represents not only a biological process but also a critical clinical barrier to curative cancer treatment.
Future research must prioritize viewing EMT as a dynamic and reversible spectrum rather than a binary switch. Key directions include developing reliable biomarkers to capture EMT states, tracking transitions in real time, and designing therapies that selectively target EMT pathways without disrupting normal regenerative processes. Integrating EMT-directed strategies with immunotherapy, chemotherapy, or targeted agents could provide a path to overcoming resistance and preventing metastatic progression.
As precision oncology advances, unraveling the complexities of EMT will be essential to crafting personalized, more effective treatment regimens. Ultimately, targeting EMT holds the promise not only of halting disease progression but also of improving long-term survival and quality of life for cancer patients.
The clinical implications of EMT in cancer are profound, as this process fundamentally reshapes tumor behavior, prognosis, and therapeutic response. A key outcome of EMT is its role in metastasis, the leading cause of cancer-related mortality. By enabling epithelial tumor cells to acquire mesenchymal traits, EMT promotes detachment from the primary tumor, invasion into surrounding tissues, intravasation into blood or lymphatic vessels, and colonization of distant organs. This transition also enhances CTC survival under mechanical and immune stress, thereby increasing metastatic efficiency. Clinically, tumors with mesenchymal features or EMT marker expression are often associated with advanced stage, poor differentiation, higher recurrence rates, and reduced overall survival. The detection of EMT signatures in tissue biopsies or liquid biopsies, such as CTCs and circulating miRNAs, is therefore gaining traction as a prognostic tool and an avenue for early identification of aggressive disease. Moreover, assessing EMT status may enable more precise patient stratification and better prediction of metastatic potential, guiding tailored surveillance and intervention strategies.
Another significant clinical consequence of EMT is its contribution to therapeutic resistance, which undermines the efficacy of chemotherapy, radiotherapy, and targeted agents. EMT-driven cancer cells frequently resist treatment by reducing apoptotic sensitivity, altering cell-cycle dynamics, increasing drug efflux, and enriching for CSC populations. In breast, lung, and PC, EMT has been linked to poor responses to cisplatin, gemcitabine, and TKIs, limiting the effectiveness of first-line regimens and narrowing options for salvage therapies. Clinically, this highlights the importance of integrating EMT biomarkers into diagnostic and therapeutic frameworks.
Emerging strategies to counteract EMT include agents that reverse or modulate EMT pathways, such as TGF-β inhibitors, Wnt pathway modulators, and miRNA-based therapeutics, currently being tested in preclinical and clinical studies. Combining these approaches with existing treatments holds promise for overcoming resistance and achieving more durable responses. As EMT continues to be recognized as a central node in cancer biology, its integration into clinical decision-making could transform patient management, improving both short-term outcomes and long-term survival.
Within the TME, EMT arises from a highly interconnected signaling network where extracellular cues, including TGF-β, Wnt/β-catenin, Notch ligands, HGF/c-Met, IGF, inflammatory cytokines such as IL-6 and TNF-α, and hypoxia converge on EMT transcription factors (Snail, Slug, Twist, ZEB1/2) to induce and stabilize mesenchymal phenotypes. Canonical TGF-β/SMAD signaling directly activates EMT-TF transcription, while non-canonical branches (PI3K/Akt/mTOR, MAPK/ERK, Rho-GTPases/JNK) remodel junctions, reorganize cytoskeletal architecture, and promote survival under stress. Wnt/β-catenin not only induces Snail expression but also stabilizes it through the Axin2-GSK3β-Snail axis, reinforcing E-cadherin repression. Notch signaling activates NF-κB, driving the expression of Snail, Twist, and ZEB, while simultaneously amplifying cytokine secretion that sustains EMT. Similarly, IL-6/JAK/STAT3 signaling enhances ZEB1 expression and cooperates with the PI3K/Akt and MAPK pathways. In contrast, hypoxia, via HIF-1/2, enhances ZEB1 and promotes metabolic reprogramming.
Tumor-intrinsic alterations further shape these pathways: PTEN loss intensifies PI3K/Akt and β-catenin activity; RAS/MAPK activation synergizes with PTEN deficiency to drive aggressive EMT and metastasis; and ATM-mediated JAK/STAT signaling links EMT to PD-L1 expression and immune evasion. Crosstalk with stromal elements compounds these effects: CAFs and stellate cells release Wnt ligands and HGF, while exosomal miRNAs from mesenchymal cells activate STAT3 in neighboring tumor cells. Reciprocal feedback loops reinforce this circuitry, for example, EMT-TFs repress the miR-200 family to sustain ZEB expression, while ZEB1 strengthens STAT3-driven programs. ECM remodeling by MMPs and integrin signaling integrates with these pathways to enhance invasion. Partial EMT states further complicate the picture, as cells retain epithelial traits for collective migration yet acquire mesenchymal motility, stemness, and drug resistance.
The outcome of this signaling web is enhanced metastasis, immune evasion, and multidrug resistance, exemplified by c-Met/PI3K/Akt-mediated gemcitabine resistance, STAT3-driven cisplatin resistance, and Wnt/β-catenin-linked chemoresistance. EMT thus functions less as a linear cascade than as a web of interlocking feedback and feed-forward loops. This complexity suggests that effective therapeutic strategies will require simultaneous targeting of multiple nodes, such as co-inhibition of TGF-β and PI3K/Akt, or dual blockade of STAT3 with c-Met or Wnt, to collapse EMT plasticity and re-sensitize tumors to treatment.
EMT-related pathways can be harnessed for both diagnosis and prognosis by converting their molecular footprints into measurable tissue- and liquid-biopsy biomarkers. Tumors exhibiting mesenchymal features or EMT marker expression, such as cadherin switching, vimentin upregulation, and loss of E-cadherin, consistently correlate with advanced disease stage, higher recurrence rates, and poorer survival outcomes. Detecting EMT signatures in biopsies or, non-invasively, in CTCs and EMT-associated miRNAs, enables early identification of high-risk patients and can guide decisions on intensive surveillance or, in some contexts, therapy selection.
Pathway-level readouts of signaling activity, including TGF-β, Wnt/β-catenin, Notch, PI3K/Akt/mTOR, and STAT3, together with downstream EMT transcription factors such as Snail, Slug, Twist, and ZEB1/2, provide an additional layer of precision. These signatures reflect the upstream circuitry that drives invasion, immune evasion, and plasticity, enabling multi-analyte panels to detect partial EMT states that underlie heterogeneity and poor clinical outcomes. Epigenetic and transcriptional consequences of EMT signaling, such as TGF-β-induced DNA methylation and chromatin remodeling at adhesion and cytoskeletal genes, provide more stable diagnostic anchors and may explain the persistent prognostic differences observed across cancers. For example, the cadherin switch in cholangiocarcinoma is associated with inferior survival and can be incorporated into pathology assessments.
In practice, integrating EMT pathway activation profiles with liquid-biopsy metrics (CTC phenotype, EMT-linked miRNAs) can help predict metastatic potential and resistance to agents such as cisplatin, gemcitabine, and TKIs. Such information supports the early use of combination regimens or referral to clinical trials in patients at high risk of therapeutic failure. Moreover, because EMT lies at the center of TME crosstalk, including cytokine, hypoxia, and stromal signals, composite EMT pathway scores show promise as prognostic and treatment triage tools. Serial measurement of these scores can also serve as pharmacodynamic markers in trials testing TGF-β or Wnt modulators, PI3K/Akt inhibitors, or miRNA-based strategies designed to reverse EMT and improve outcomes.
Partial EMT describes an intermediate state in which cancer cells acquire mesenchymal properties, such as motility, invasiveness, and stemness, while still retaining key epithelial features, including cell-cell adhesion. This hybrid phenotype profoundly impacts tumor progression, intratumoral heterogeneity, and therapy resistance. Unlike full EMT, in which cells fully adopt a mesenchymal identity, partial EMT allows a continuum of phenotypes that confer on tumor cells the plasticity to dynamically adapt to microenvironmental pressures. This adaptability supports collective migration, whereby clusters of partially transitioned cells disseminate together, maintaining intercellular junctions for survival while possessing sufficient mesenchymal traits to invade, extravasate, and colonize distant sites more effectively than fully mesenchymal single cells.
At the molecular level, partial EMT is sustained by finely balanced signaling from pathways such as TGF-β, Wnt/β-catenin, Notch, and PI3K/Akt, in conjunction with transcriptional regulators, including Snail, Twist, and ZEB proteins, which are often expressed at intermediate levels or in oscillatory patterns. Feedback with the miR-200 family and other noncoding RNAs further stabilizes this state. Microenvironmental cues, including hypoxia, inflammatory cytokines, stromal fibroblasts, and immune cells, reinforce reversible and context-dependent EMT programs.
Clinically, partial EMT is associated with poor prognosis across multiple cancers, as it drives metastatic dissemination, immune evasion, and chemoresistance, while generating intratumoral heterogeneity that complicates therapeutic response. Biomarkers that capture this hybrid state, such as the co-expression of E-cadherin with vimentin or N-cadherin, along with transcriptional signatures of partial EMT, offer diagnostic and prognostic value. From a therapeutic perspective, targeting partial EMT presents both challenges and opportunities: its plasticity underpins metastatic efficiency but also exposes vulnerabilities in adhesion, collective migration, and regulatory feedback loops. Thus, modulating partial EMT rather than enforcing complete EMT reversal may represent a more effective strategy in cancer treatment.
Targeting EMT through CRISPR-based genome editing and drug repurposing has emerged as a promising strategy to combat metastasis, immune evasion, and therapy resistance by directly dismantling the molecular circuitry underlying phenotypic plasticity. CRISPR enables precise modulation of EMT-associated genes, such as the transcription factors Snail, Slug, Twist, and ZEB1/2, as well as upstream regulators such as TGF-β receptors, AXL, and PI3K/Akt components, offering the potential to reverse mesenchymal traits, restore epithelial adhesion, and enhance immune recognition. Beyond gene knockouts, CRISPR can be harnessed for gene activation or epigenome editing to reprogram EMT-suppressive pathways, reinstate miR-200 family expression, or restore antigen presentation via MHC class I, thereby sensitizing tumors to immunotherapy.
Drug repurposing offers a complementary and accelerated pathway to clinical application by identifying EMT-modulating properties in FDA-approved agents. Examples include metformin’s inhibition of TGF-β/Smad signaling, statins’ suppression of the Rho/ROCK pathways, and COX-2 inhibitors’ disruption of MDSC-mediated EMT induction, which are readily available tools to blunt EMT-driven metastasis and resistance. Combining CRISPR-based gene targeting with repurposed drugs may yield synergistic effects, for instance, disabling EMT-TFs with CRISPR while simultaneously blocking compensatory signaling through repurposed kinases or anti-inflammatory agents, collectively restraining plasticity and metastatic potential.
Crucially, these strategies enable precise targeting of EMT nodes without broadly disrupting normal epithelial physiology. When integrated with immunotherapy or chemotherapy, CRISPR and drug repurposing approaches could redefine EMT from a major driver of cancer lethality into a tractable therapeutic vulnerability.
Translating EMT biology into the clinic presents several interconnected challenges. First, EMT is not a simple binary switch but a dynamic spectrum that includes hybrid or partial EMT states, which drive intratumoral heterogeneity, immune evasion, and therapy resistance. This plasticity complicates diagnostics, trial enrollment, and drug targeting, as single-marker assays often fail to capture transient states, underscoring the need for tools that track EMT transitions in real time, validate composite biomarkers, and distinguish malignant EMT from normal regenerative programs to avoid on-target toxicity.
Therapeutically, pathway redundancy and extensive crosstalk with the TME via CAF- and stellate-derived ligands, exosomal miRNAs, and ECM-integrin signaling create feedback loops that rapidly circumvent single-node inhibitors. This suggests that rational drug combinations (co-inhibiting TGF-β and PI3K/Akt, or pairing STAT3 blockade with c-Met or Wnt modulation), ideally in combination with immunotherapy or chemotherapy, will be necessary to disrupt EMT plasticity and restore tumor sensitivity.
Drug delivery remains a significant challenge: many EMT-targeted agents, such as siRNAs, miRNA mimetics, and natural products, exhibit poor bioavailability and inadequate tumor targeting. Nanoparticle-based platforms capable of delivering siTWIST or similar cargos, as well as adjunct approaches such as therapeutic ultrasound, to enhance intratumoral accumulation, show promise in overcoming these barriers. Adding to the complexity, EMT pathways are highly context-dependent; for example, Notch signaling can function as either an oncogene or a tumor suppressor, depending on the cellular lineage and state. This highlights the importance of refined patient selection and adaptive, pathway-aware treatment strategies rather than uniform inhibitor approaches.
Looking ahead, key priorities include developing robust EMT signatures in tissue and liquid biopsies to capture hybrid states and predict metastasis or resistance; embedding EMT readouts into precision oncology workflows to guide surveillance, neoadjuvant therapy, and rational combinations; and advancing next-generation delivery systems to engage EMT drivers in patients effectively. Together, the maturation of biomarkers, context-aware combination strategies, and more innovative delivery platforms could shift EMT from a conceptual vulnerability to a practical therapeutic target that improves prognosis and response durability across solid tumors.
References
1. Siegel RL, Kratzer TB, Giaquinto AN, Sung H, Jemal A. Cancer statistics, 2025. CA Cancer J Clin. 2025;75(1):10-45. https://doi.org/10.3322/caac.21871
2. Wagle NS, Nogueira L, Devasia TP, Mariotto AB, Yabroff KR, Islami F, et al. Cancer treatment and survivorship statistics, 2025. CA Cancer J Clin. 2025;75(4):308-340. https://doi.org/10.3322/caac.70011.
3. Nguyen DX, Massagué J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007; 8(5):341‑52. https://doi.org/10.1038/nrg2101
4. Chen LL, Blumm N, Christakis NA, Barabási A-L, Deisboeck TS. Cancer metastasis networks and the prediction of progression patterns. Br J Cancer. 2009;101(5):749‑58. https://doi.org/10.1038/sj.bjc.6605214
5. Ganesh K, Massagué J. Targeting metastatic cancer. Nat Med. 2021;27(1):34-44. https://doi.org/10.1038/s41591-020-01195-4
6. Smith BN, Bhowmick NA. Role of EMT in Metastasis and Therapy Resistance. J Clin Med. 2016;5(2):17. https://doi.org/10.3390/jcm5020017
7. Santamaria PG, Moreno‑Bueno G, Portillo F, Cano A. EMT: present and future in clinical oncology. Mol Oncol. 2017;(7):718‑738. https://doi.org/10.1002/1878-0261.12091
8. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;(2):97‑110. https://doi.org/10.1038/nrc3447
9. Díaz‑López A, Moreno‑Bueno G, Cano A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag Res. 2014;6:205‑216. https://doi.org/10.2147/cmar.s38156
10. Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347‑376. https://doi.org/10.1146/annurev-cellbio-092910-154036
11. Nieto MA, Cano A. The epithelial‑mesenchymal transition under control: global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012;(5):361‑368. https://doi.org/10.1016/j.semcancer.2012.05.003
12. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415‑428. https://doi.org/10.1038/nrc2131
13. Tam WL, Weinberg RA. The epigenetics of epithelial‑mesenchymal plasticity in cancer. Nat Med. 2013;19(11):1438‑1449. https://doi.org/10.1038/nm.3336
14. Yang J, Weinberg RA. Epithelial‑mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008 Jun;14(6):818‑829. https://doi.org/10.1016/j.devcel.2008.05.009
15. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial‑mesenchymal transitions in development and disease. Cell. 2009;139(5):871‑890. https://doi.org/10.1016/j.cell.2009.11.007
16. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21‑45. https://doi.org/10.1016/j.cell.2016.06.028
17. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017 Feb 9;168(4):670‑691. https://doi.org/10.1016/j.cell.2016.11.037
18. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342(6163):1234850. https://doi.org/10.1126/science.1234850
19. Brabletz S, Schuhwerk H, Brabletz T, Stemmler MP. Dynamic EMT: a multi‑tool for tumor progression. EMBO J. 2021;40(8):e108647. https://doi.org/10.15252/embj.2021108647
20. Hay ED. An overview of epithelio‑mesenchymal transformation. Cells Tissues Organs. 1995;154(1):8‑20. https://doi.org/10.1159/000147748
21. Arnoux V, Nassour M, L’Helgoualc’h A, Hipskind RA, Savagner P. Erk5 controls Slug expression and keratinocyte activation during wound healing. Mol Biol Cell. 2008;19(12):4738‑4749. https://doi.org/10.1091/mbc.e07-10-1078
22. Ahmed N, Maines‑Bandiera S, Quinn MA, Unger WG, Dedhar S, Auersperg N. Molecular pathways regulating EGF‑induced epithelio‑mesenchymal transition in human ovarian surface epithelium. Am J Physiol Cell Physiol. 2006;290(6):C1532‑C1542. https://doi.org/10.1152/ajpcell.00478.2005
23. Kalluri R. EMT: when epithelial cells decide to become mesenchymal‑like cells. J Clin Invest. 2009;119(6):1417‑1419. https://doi.org/10.1172/jci39675
24. Gumbiner BM. Epithelial morphogenesis. Cell. 1992;69(3):385‑387. https://doi.org/10.1016/0092-8674(92)90440-n
25. Yeaman C, Grindstaff KK, Hansen MD, Nelson WJ. Cell polarity: versatile scaffolds keep things in place. Curr Biol. 1999;9(13):R515‑R517. https://doi.org/10.1016/s0960-9822(99)80324-8
26. Kalluri R, Weinberg RA. The basics of epithelial‑mesenchymal transition. J Clin Invest. 2009;119(6):1420‑1428. https://doi.org/10.1172/jci39104
27. Felipe Lima J, Nofech‑Mozes S, Bayani J, Bartlett JMS. EMT in breast carcinoma—a review. J Clin Med. 2016;5(7):65. https://doi.org/10.3390/jcm5070065
28. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265‑273. https://doi.org/10.1038/nrc2620
29. Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial‑mesenchymal transition induced by transforming growth factor‑beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15(2):169‑190. https://doi.org/10.1007/s10911-010-9181-1
30. Singh M, Yelle N, Venugopal C, Singh SK. EMT: Mechanisms and therapeutic implications. Pharmacol Ther. 2018;182:80‑94. https://doi.org/10.1016/j.pharmthera.2017.08.009.
31. Heerboth S, Housman G, Leary M, Longacre M, Byler S, Lapinska K, et al. EMT and tumor metastasis. Clin Transl Med. 2015;4:6. https://doi.org/10.1186/s40169-015-0048-3
32. Boutet A, Esteban MA, Maxwell PH, Nieto MA. Reactivation of Snail genes in renal fibrosis and carcinomas: a process of reversed embryogenesis? Cell Cycle. 2007;6(6):638‑642. https://doi.org/10.4161/cc.6.6.4022
33. Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, van Roy F, Lee-Kwon W, et al. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003;63(9):2172-2178. PMID: 12727836
34. Vega S, Morales AV, Ocana OH, Valdés F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18(10):1131‑1143. https://doi.org/10.1101/gad.294104
35.Kim HJ, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol. 2007;27(8):3165‑3175. https://doi.org/10.1128/mcb.01315-06
36. Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, et al. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68(7):2479‑2488. https://doi.org/10.1158/0008-5472.can-07-2559
37. Xu J, Liu S, Yang X, Cao S, Zhou Y. Paracrine HGF promotes EMT and mediates the effects of PSC on chemoresistance by activating c-Met/PI3K/Akt signaling in pancreatic cancer in vitro. Life Sci. 2020;263:118523. https://doi.org/10.1016/j.lfs.2020.118523
38. Zhang B, Li Y, Wu Q, Xie L, Barwick B, Fu C, et al. Acetylation of KLF5 maintains EMT and tumorigenicity to cause chemoresistant bone metastasis in prostate cancer. Nat Commun. 2021;12(1):1714. https://doi.org/10.1038/s41467-021-21976-w
39. Mitra T, Prasad P, Mukherjee P, Chaudhuri SR, Chatterji U, Roy SS. Stemness and chemoresistance are imparted to the OC cells through TGFβ1 driven EMT. J Cell Biochem. 2018;119(7):5775‑5787. https://doi.org/10.1002/jcb.26753
40. Meng X, Xiao W, Sun J, Li W, Yuan H, Yu T, et al. CircPTK2/PABPC1/SETDB1 axis promotes EMT-mediated tumor metastasis and gemcitabine resistance in bladder cancer. Cancer Lett. 2023;554:216023. https://doi.org/10.1016/j.canlet.2022.216023
41. Zhang L, Zhao XL, Cao ZJ, Li KD, Xu LY, Tang F, et al. Ginsenoside CK inhibits EMT and overcomes oxaliplatin resistance in gastric cancer by targeting the PI3K/Akt pathway. Phytomedicine. 2025;140:156516. https://doi.org/10.1016/j.phymed.2025.156516
42. Meng Q, Shi S, Liang C, Liang D, Hua J, Zhang B, et al. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3β/Snail signaling. Oncogene. 2018;37(44):5843‑5857. https://doi.org/10.1038/s41388-018-0392-z
43. Li Y, Hong J, Jung BK, Oh E, Yun CO. Oncolytic Ad co-expressing decorin and Wnt decoy receptor overcomes chemoresistance of desmoplastic tumor through degradation of ECM and inhibition of EMT. Cancer Lett. 2019;459:15‑29. https://doi.org/10.1016/j.canlet.2019.05.033
44. Zhu W, Sun J, Jing F, Xing Y, Luan M, Feng Z, et al. GLI2 inhibits cisplatin sensitivity in gastric cancer through DEC1/ZEB1 mediated EMT. Cell Death Dis. 2025;16(1):204. https://doi.org/10.1038/s41419-025-07564-6
45. Xiong Y, Sun F, Dong P, Watari H, Yue J, Yu MF, et al. iASPP induces EMT and cisplatin resistance in human cervical cancer through miR-20a-FBXL5/BTG3 signaling. J Exp Clin Cancer Res. 2017;36(1):48. https://doi.org/10.1186/s13046-017-0520-6
46. Weadick B, Nayak D, Persaud AK, Hung SW, Raj R, Campbell MJ, Chen W, Li J, Williams TM, Govindarajan R. EMT‑induced gemcitabine resistance in pancreatic cancer involves the functional loss of equilibrative nucleoside transporter 1. Mol Cancer Ther. 2021;20(2):410‑422. https://doi.org/10.1158/1535-7163.mct-20-0316
47. Zhao H, Duan Q, Zhang Z, Li H, Wu H, Shen Q, et al. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J Cell Mol Med. 2017;21(9):2055‑2067. https://doi.org/10.1111/jcmm.13126
48. Okada Y, Takahashi N, Takayama T, Goel A. LAMC2 promotes cancer progression and gemcitabine resistance through modulation of EMT and ATP-binding cassette transporters in pancreatic ductal adenocarcinoma. Carcinogenesis. 2021;42(4):546‑556. https://doi.org/10.1093/carcin/bgab011
49. Zhang PF, Li KS, Shen YH, Gao PT, Dong ZR, Cai JB, et al. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 2016;7(4):e2201. https://doi.org/10.1038/cddis.2015.324
50. Maseki S, Ijichi K, Tanaka H, Fujii M, Hasegawa Y, Ogawa T, et al. Acquisition of EMT phenotype in the gefitinib-resistant cells of a head and neck squamous cell carcinoma cell line through Akt/GSK-3β/snail signalling pathway. Br J Cancer. 2012;106(6):1196‑1204. https://doi.org/10.1038/bjc.2012.24
51. Sale MJ, Balmanno K, Saxena J, Ozono E, Wojdyla K, McIntyre RE, et al. MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAF(V600E) amplification whereas KRAS(G13D) amplification promotes EMT-chemoresistance. Nat Commun. 2019;10(1):2030. https://doi.org/10.1038/s41467-019-09438-w
52. Sreekumar R, Al-Saihati H, Emaduddin M, Moutasim K, Mellone M, Patel A, et al. The ZEB2-dependent EMT transcriptional programme drives therapy resistance by activating nucleotide excision repair genes ERCC1 and ERCC4 in colorectal cancer. Mol Oncol. 2021;15(8):2065-2083. https://doi.org/10.1002/1878-0261.12965
53.Wang C, Li A, Yang S, Qiao R, Zhu X, Zhang J. CXCL5 promotes mitomycin C resistance in non-muscle invasive bladder cancer by activating EMT and NF-κB pathway. Biochem Biophys Res Commun. 2018;498:862‑868. https://doi.org/10.1016/j.bbrc.2018.03.071
54. Sun J, Xu Z, Lv H, Wang Y, Wang L, Ni Y, Wang X, Hu C, Chen S, Teng F, Chen W, Cheng X. eIF5A2 regulates the resistance of gastric cancer cells to cisplatin via induction of EMT. Am J Transl Res. 2018;10(12):4269‑4279. PMID: 30662669. PMCID: PMC6325524
55. Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, Geraci J, et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer. 2012;12:91. https://doi.org/10.1186/1471-2407-12-91
56. Morelli AP, Tortelli TC Jr, Mancini MCS, Pavan ICB, Silva LGS, Severino MB, et al. STAT3 contributes to cisplatin resistance, modulating EMT markers, and the mTOR signaling in lung adenocarcinoma. Neoplasia. 2021;23(10):1048‑1058. https://doi.org/10.1016/j.neo.2021.08.003
57. Chen K, Xu J, Tong YL, Yan JF, Pan Y, Wang WJ, et al. Rab31 promotes metastasis and cisplatin resistance in stomach adenocarcinoma through Twist1-mediated EMT. Cell Death Dis. 2023;14(2):115. https://doi.org/10.1038/s41419-023-05596-4
58. Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding Q, et al. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 2019;38(1):149. https://doi.org/10.1186/s13046-019-1161-8
59. Sinha S, Sharma S, Sharma A, Vora J, Shrivastava N. Sulforaphane-cisplatin combination inhibits the stemness and metastatic potential of TNBCs via down regulation of sirtuins-mediated EMT signaling axis. Phytomedicine. 2021;84:153492. https://doi.org/10.1016/j.phymed.2021.153492
60. Liang F, Ren C, Wang J, Wang S, Yang L, Han X, et al. The crosstalk between STAT3 and p53/RAS signaling controls cancer cell metastasis and cisplatin resistance via the Slug/MAPK/PI3K/AKT-mediated regulation of EMT and autophagy. Oncogenesis. 2019;8(10):59. https://doi.org/10.1038/s41389-019-0165-8
61. Xu T, Zhang J, Chen W, Pan S, Zhi X, Wen L, et al. ARK5 promotes doxorubicin resistance in hepatocellular carcinoma via epithelial-mesenchymal transition. Cancer Lett. 2016;377(2):140‑148. https://doi.org/10.1016/j.canlet.2016.04.026.
62. Paramanantham A, Jung EJ, Kim HJ, Jeong BK, Jung JM, Kim GS, et al. Doxorubicin‑resistant TNBC cells exhibit rapid growth with cancer stem cell‑like properties and EMT phenotype, which can be transferred to parental cells through autocrine signaling. Int J Mol Sci. 2021;22(22):12438. https://doi.org/10.3390/ijms222212438
63. Wang S, Yan Y, Cheng Z, Hu Y, Liu T. Sotetsuflavone suppresses invasion and metastasis in non-small-cell lung cancer A549 cells by reversing EMT via the TNF-α/NF-κB and PI3K/AKT signaling pathway. Cell Death Discov. 2018;4:26. https://doi.org/10.1038/s41420-018-0026-9
64. Jiao D, Wang J, Lu W, Tang X, Chen J, Mou H, et al. Curcumin inhibited HGF-induced EMT and angiogenesis through regulating c-Met dependent PI3K/Akt/mTOR signaling pathways in lung cancer. Mol Ther Oncolytics. 2016;3:16018. https://doi.org/10.1038/mto.2016.18
65. Wei R, Xiao Y, Song Y, Yuan H, Luo J, Xu W. FAT4 regulates the EMT and autophagy in colorectal cancer cells in part via the PI3K-AKT signaling axis. J Exp Clin Cancer Res. 2019;38(1):112. https://doi.org/10.1186/s13046-019-1043-0
66. Chi M, Liu J, Mei C, Shi, Y, Liu N, Jiang X, et al. TEAD4 functions as a prognostic biomarker and triggers EMT via PI3K/AKT pathway in bladder cancer. J Exp Clin Cancer Res. 2022;41(1):175. https://doi.org/10.1186/s13046-022-02377-3
67. Ha GH, Park JS, Breuer EKY. TACC3 promotes epithelial-mesenchymal transition (EMT) through the activation of PI3K/Akt and ERK signaling pathways. Cancer Lett. 2013;332(1):63‑73. https://doi.org/10.1016/j.canlet.2013.01.013.
68. Wang SC, Chai DS, Chen CB, Wang ZY, Wang L. HPIP promotes thyroid cancer cell growth, migration and EMT through activating PI3K/AKT signaling pathway. Biomed Pharmacother. 2015;75:33‑39. https://doi.org/10.1016/j.biopha.2015.08.027.
69. Meng J, Zhang XT, Liu XL, Fan L, Li C, Sun, Y, et al. WSTF promotes proliferation and invasion of lung cancer cells by inducing EMT via PI3K/Akt and IL-6/STAT3 signaling pathways. Cell Signal. 2016;28(11):1673‑1682. https://doi.org/10.1016/j.cellsig.2016.07.008.
70. Cui P, Li H, Wang C, Liu Y, Zhang M, Yin Y, et al. UBE2T regulates epithelial-mesenchymal transition through the PI3K-AKT pathway and plays a carcinogenic role in ovarian cancer. J Ovarian Res. 2022;15(1):103. https://doi.org/10.1186/s13048-022-01034-9
71. Tang Z, Ding Y, Shen Q, Zhang C, Li J, Nazar M, et al. KIAA1199 promotes invasion and migration in non-small-cell lung cancer (NSCLC) via PI3K-Akt mediated EMT. J Mol Med (Berl). 2019;97(1):127‑140. https://doi.org/10.1007/s00109-018-1721-y
72. Ghobashi AH, Kimani JW, Ladaika CA, O’Hagan HM. PTEN depletion reduces H3K27me3 levels to promote epithelial-to-mesenchymal transition in epithelial colorectal cancer cells. PLoS One. 2024;19(11):e0313769. https://doi.org/10.1371/journal.pone.0313769
73. Kim J, Kang HS, Lee YJ, Lee HJ, Yun J, Shin JH, et al. EGR1-dependent PTEN upregulation by 2-benzoyloxycinnamaldehyde attenuates cell invasion and EMT in colon cancer. Cancer Lett. 2014;349(1):35‑44. https://doi.org/10.1016/j.canlet.2014.03.025.
74. Perumal E, So Youn K, Sun S, Seung-Hyun J, Suji M, Jieying L, et al C. PTEN inactivation induces epithelial-mesenchymal transition and metastasis by intranuclear translocation of β-catenin and snail/slug in non-small cell lung carcinoma cells. Lung Cancer. 2019;130:25‑34. https://doi.org/10.1016/j.lungcan.2019.01.013.
75. Gan L, Xu M, Hua R, Tan C, Zhang J, Gong Y, et al. The polycomb group protein EZH2 induces epithelial-mesenchymal transition and pluripotent phenotype of gastric cancer cells by binding to PTEN promoter. J Hematol Oncol. 2018;11(1):9. https://doi.org/10.1186/s13045-017-0547-3
76. Yokoi A, Minami M, Hashimura M, Oguri Y, Matsumoto T, Hasegawa Y, et al. PTEN overexpression and nuclear β-catenin stabilization promote morular differentiation through induction of epithelial-mesenchymal transition and cancer stem cell-like properties in endometrial carcinoma. Cell Commun Signal. 2022;20(1):181. https://doi.org/10.1186/s12964-022-00999-w
77. Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 Down-regulates PTEN Expression and Signals through PI3K to Promote Epithelial-Mesenchymal Transition. Cancer Res. 2007;67(7):2922‑2926. https://doi.org/10.1158/0008-5472.can-06-3598
78. Xie S, Lu Z, Lin Y, Shen L, Yin C. Upregulation of PTEN suppresses invasion in Tca8113 tongue cancer cells through repression of epithelial-mesenchymal transition (EMT). Tumour Biol. 2016;37(5):6681‑6689. https://doi.org/10.1007/s13277-015-4486-8
79. Bowen KA, Doan HQ, Zhou BP, Wang Q, Zhou Y, Rychahou PG, et al. PTEN loss induces epithelial—mesenchymal transition in human colon cancer cells. Anticancer Res. 2009;29(11):4439‑4449. PMID: 20032390. PMCID: PMC2932708
80. Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, et al. Pten Loss and RAS/MAPK Activation Cooperate to Promote EMT and Metastasis Initiated from Prostate Cancer Stem/Progenitor Cells. Cancer Res. 2012;72(7):1878‑1889. https://doi.org/10.1158/0008-5472.can-11-3132
81. Wang T, Lin F, Sun X, Jiang L, Mao R, Zhou S, et al. HOXB8 enhances the proliferation and metastasis of colorectal cancer cells by promoting EMT via STAT3 activation. Cancer Cell Int. 2019;19:3. https://doi.org/10.1186/s12935-018-0717-6
82. Wu YS, Chung I, Wong WF, Masamune A, Sim MS, Looi CY. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim Biophys Acta Gen Subj. 2017;1861(2):296‑306. https://doi.org/10.1016/j.bbagen.2016.10.006.
83. Ma Q, Chen F, Liu Y, Wu K, Bu Z, Qiu C, et al. Integrated transcriptomic and proteomic analysis reveals Guizhi-Fuling Wan inhibiting STAT3-EMT in ovarian cancer progression. Biomed Pharmacother. 2024;170:116016. https://doi.org/10.1016/j.biopha.2023.116016.
84. Park SJ, Jung HJ. Bufotalin Suppresses Proliferation and Metastasis of Triple-Negative Breast Cancer Cells by Promoting Apoptosis and Inhibiting the STAT3/EMT Axis. Molecules. 2023;28(19):6783. https://doi.org/10.3390/molecules28196783
85. Guo H, Hu Z, Yang X, Yuan Z, Gao Y, Chen J, et al. STAT3 inhibition enhances gemcitabine sensitivity in pancreatic cancer by suppressing EMT, immune escape and inducing oxidative stress damage. Int Immunopharmacol. 2023;123:110709. https://doi.org/10.1016/j.intimp.2023.110709.
86. Chen G, Tang N, Wang C, Xiao L, Yu M, Zhao L, et al. TNF-α-inducing protein of Helicobacter pylori induces epithelial-mesenchymal transition (EMT) in gastric cancer cells through activation of IL-6/STAT3 signaling pathway. Biochem Biophys Res Commun. 2017;484(2):311‑317. https://doi.org/10.1016/j.bbrc.2017.01.110.
87. Wang LN, Zhang ZT, Wang L, Wei HX, Zhang T, Zhang LM, et al. TGF-β1/SH2B3 axis regulates anoikis resistance and EMT of lung cancer cells by modulating JAK2/STAT3 and SHP2/Grb2 signaling pathways. Cell Death Dis. 2022;13(5):472. https://doi.org/10.1038/s41419-022-04890-x
88. Xiong H, Hong J, Du W, Lin YW, Ren LL, Wang YC, et al. Roles of STAT3 and ZEB1 Proteins in E-cadherin Down-regulation and Human Colorectal Cancer Epithelial-Mesenchymal Transition. J Biol Chem. 2012;287(8):5819‑5832. https://doi.org/10.1074/jbc.M111.295964.
89. Zhang X, Sai B, Wang F, Wang L, Wang Y, Zheng L, et al. Hypoxic BMSC-derived exosomal miRNAs promote metastasis of lung cancer cells via STAT3-induced EMT. Mol Cancer. 2019;18(1):40. https://doi.org/10.1186/s12943-019-0959-5
90. Lin WH, Chang YW, Hong MX, Hsu TC, Lee KC, Lin C, et al. STAT3 phosphorylation at Ser727 and Tyr705 differentially regulates the EMT-MET switch and cancer metastasis. Oncogene. 2021;40(4):791‑805. https://doi.org/10.1038/s41388-020-01566-8
91. Ahmad A, Sarkar SH, Bitar B, Ali S, Aboukameel A, Sethi S, et al. Garcinol Regulates EMT and Wnt Signaling Pathways In Vitro and In Vivo, Leading to Anticancer Activity against Breast Cancer Cells. Mol Cancer Ther. 2012;11(10):2193‑2201. https://doi.org/10.1158/1535-7163.mct-12-0232-t
92. Jiang L, Yang YD, Fu L, Xu W, Liu D, Liang Q, et al. CLDN3 inhibits cancer aggressiveness via Wnt-EMT signaling and is a potential prognostic biomarker for hepatocellular carcinoma. Oncotarget. 2014;5(17):7663‑7676. https://doi.org/10.18632/oncotarget.2288
93. Li Q, Lai Q, He C, Fang Y, Yan Q, Zhang Y, et al. RUNX1 promotes tumour metastasis by activating the Wnt/β-catenin signalling pathway and EMT in colorectal cancer. J Exp Clin Cancer Res. 2019;38(1):334. https://doi.org/10.1186/s13046-019-1330-9
94. Yang S, Liu Y, Li MY, Ng CSH, Yang SL, Wang S, et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/β-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. 2017;16(1):124. https://doi.org/10.1186/s12943-017-0700-1
95. Tang Q, Chen J, Di Z, Yuan W, Zhou Z, Liu Z, et al. TM4SF1 promotes EMT and cancer stemness via the Wnt/β-catenin/SOX2 pathway in colorectal cancer. J Exp Clin Cancer Res. 2020;39(1):232. https://doi.org/10.1186/s13046-020-01690-z
96. Mosa MH, Michels BE, Menche C, Nicolas AM, Darvishi T, Greten FR, et al. A Wnt-Induced Phenotypic Switch in Cancer-Associated Fibroblasts Inhibits EMT in Colorectal Cancer. Cancer Res. 2020;80(24):5569‑5582. https://doi.org/10.1158/0008-5472.can-20-0263
97. Ma X, Wang B, Wang X, Luo Y, Fan W. NANOGP8 is the key regulator of stemness, EMT, Wnt pathway, chemoresistance, and other malignant phenotypes in gastric cancer cells. PLoS One. 2018;13(4):e0192436. https://doi.org/10.1371/journal.pone.0192436
98. Jia S, Qu T, Wang X, Feng M, Yang Y, Feng X, et al. KIAA1199 promotes migration and invasion by Wnt/β-catenin pathway and MMPs mediated EMT progression and serves as a poor prognosis marker in gastric cancer. PLoS One. 2017;12(4):e0175058. https://doi.org/10.1371/journal.pone.0175058
99. Hu W, Wang Z, Zhang S, Lu X, Wu J, Yu K, et al. IQGAP1 promotes pancreatic cancer progression and epithelial-mesenchymal transition (EMT) through Wnt/β-catenin signaling. Sci Rep. 2019;9(1):7539. https://doi.org/10.1038/s41598-019-44048-y
100. Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, et al. A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8(12):1398‑1406. https://doi.org/10.1038/ncb1508
101. Liu F, Xia Z, Zhang M, Ding J, Feng Y, Wu J, et al. SMARCAD1 Promotes Pancreatic Cancer Cell Growth and Metastasis through Wnt/β-catenin-Mediated EMT. Int J Biol Sci. 2019;15(3):636‑646. https://doi.org/10.7150/ijbs.29562
102. Li H, Wang Z, Zhang W, Qian K, Liao G, Xu W, et al. VGLL4 inhibits EMT in part through suppressing Wnt/β-catenin signaling pathway in gastric cancer. Med Oncol. 2015;32(3):83. https://doi.org/10.1007/s12032-015-0539-5
103. Basu B, Karmakar S, Basu M, Ghosh MK. USP7 imparts partial EMT state in colorectal cancer by stabilizing the RNA helicase DDX3X and augmenting Wnt/β-catenin signaling. Biochim Biophys Acta Mol Cell Res. 2023;1870(4):119446. https://doi.org/10.1016/j.bbamcr.2023.119446
104. Xie SL, Fan S, Zhang SY, Chen WX, Li QX, Pan GK, et al. SOX8 regulates cancer stem-like properties and cisplatin-induced EMT in tongue squamous cell carcinoma by acting on the Wnt/β-catenin pathway. Int J Cancer. 2018;142(6):1252‑1265. https://doi.org/10.1002/ijc.31134.
105. DiMeo TA, Anderson K, Phadke P, Feng C, Perou CM, Naber S, et al. A Novel Lung Metastasis Signature Links Wnt Signaling with Cancer Cell Self-Renewal and Epithelial-Mesenchymal Transition in Basal-like Breast Cancer. Cancer Res. 2009;69(13):5364‑5373. https://doi.org/10.1158/0008-5472.can-08-4135
106. Duan H, Yan Z, Chen W, Wu Y, Han J, Guo H, et al. TET1 inhibits EMT of ovarian cancer cells through activating Wnt/β‑catenin signaling inhibitors DKK1 and SFRP2. Gynecol Oncol. 2017;147(2):408‑417. https://doi.org/10.1016/j.ygyno.2017.08.010.
107. Gu Y, Wang Q, Guo K, Qin W, Liao W, Wang S, et al. TUSC3 promotes colorectal cancer progression and epithelial‑mesenchymal transition (EMT) through WNT/β‑catenin and MAPK signalling. J Pathol. 2016;239(1):60‑71. https://doi.org/10.1002/path.4697.
108. Kang AR, Kim JL, Kim Y, Kang S, Oh SC, Park JK. A novel RIP1‑mediated canonical WNT signaling pathway that promotes colorectal cancer metastasis via β‑catenin stabilization‑induced EMT. Cancer Gene Ther. 2023;30(10):1403‑1413. https://doi.org/10.1038/s41417-023-00647-6
109. Shin SY, Rath O, Zebisch A, Choo SM, Kolch W, Cho KH. Functional roles of multiple feedback loops in extracellular signal‑regulated kinase and Wnt signaling pathways that regulate epithelial‑mesenchymal transition. Cancer Res. 2010;70(17):6715‑6724. https://doi.org/10.1158/0008-5472.can-10-1377
110. Li Y, Liu R, Han X, Xu W, Liu Y. PLAGL2 increases adriamycin resistance and EMT in breast cancer cells by activating the Wnt pathway. Genes Genomics. 2023;45(1):49‑57. https://doi.org/10.1007/s13258-022-01330-0
111. Chen Z, Wu W, Huang Y, Xie L, Li Y, Chen H, et al. RCC2 promotes breast cancer progression through regulation of Wnt signaling and inducing EMT. J Cancer. 2019;10(27):6837‑6847. https://doi.org/10.7150/jca.36430
112. Li Y, Liu C, Zhang X, Huang X, Liang S, Xing F, et al. CCT5 induces epithelial‑mesenchymal transition to promote gastric cancer lymph node metastasis by activating the Wnt/β‑catenin signalling pathway. Br J Cancer. 2022;126(12):1684‑1694. https://doi.org/10.1038/s41416-022-01747-0
113. Sun L, Shi C, Liu S, Zhang E, Yan L, Ji C, et al. Overexpression of NuSAP1 is predictive of an unfavourable prognosis and promotes proliferation and invasion of triple‑negative breast cancer cells via the Wnt/β‑catenin/EMT signalling axis. Gene. 2020;747:144657. https://doi.org/10.1016/j.gene.2020.144657.
114. Yang N, Hui L, Wang Y, Yang H, Jiang X. Overexpression of SOX2 promotes migration, invasion, and epithelial‑mesenchymal transition through the Wnt/β‑catenin pathway in laryngeal cancer Hep‑2 cells. Tumour Biol. 2014;35(8):7965‑7973. https://doi.org/10.1007/s13277-014-2045-3
115. Zhang X, Li H, Wang Y, Zhao H, Wang Z, Chan FL. Nuclear receptor NURR1 functions to promote stemness and epithelial‑mesenchymal transition in prostate cancer via its targeting of Wnt/β‑catenin signaling pathway. Cell Death Dis. 2024;15(3):234. https://doi.org/10.1038/s41419-024-06621-w
116. Sohn SH, Kim B, Sul HJ, Kim YJ, Kim HS, Kim H, et al. INC280 inhibits Wnt/β‑catenin and EMT signaling pathways and induces apoptosis in diffuse gastric cancer positive for c‑MET amplification. BMC Res Notes. 2019;12(1):125. https://doi.org/10.1186/s13104-019-4163-x
117. Shao M, Wang L, Zhang Q, Wang T, Wang S. STMN2 overexpression promotes cell proliferation and EMT in pancreatic cancer mediated by WNT/β‑catenin signaling. Cancer Gene Ther. 2023;30(3):472‑480. https://doi.org/10.1038/s41417-022-00568-w
118. Zhang LZ, Huang LY, Huang AL, Liu JX, Yang F. CRIP1 promotes cell migration, invasion and epithelial‑mesenchymal transition of cervical cancer by activating the Wnt/β‑catenin signaling pathway. Life Sci. 2018;207:420‑427. https://doi.org/10.1016/j.lfs.2018.05.054.
119. Wang H, Deng G, Ai M, Xu Z, Mou T, Yu J, et al. Hsp90ab1 stabilizes LRP5 to promote epithelial‑mesenchymal transition via activating AKT and Wnt/β‑catenin signaling pathways in gastric cancer progression. Oncogene. 2019;38(9):1489‑1507. https://doi.org/10.1038/s41388-018-0532-5
120. Zhou Q, Chen S, Lu M, Luo Y, Wang G, Xiao Y, et al. EFEMP2 suppresses epithelial‑mesenchymal transition via Wnt/β‑catenin signaling pathway in human bladder cancer. Int J Biol Sci. 2019;15(10):2139‑2155. https://doi.org/10.7150/ijbs.35541
121. Sun J, Yang X, Zhang R, Liu S, Gan X, Xi X, et al. GOLPH3 induces epithelial‑mesenchymal transition via Wnt/β‑catenin signaling pathway in epithelial ovarian cancer. Cancer Med. 2017;6(4):834‑844. https://doi.org/10.1002/cam4.1040
122. Ali A, Wang Z, Fu J, Ji L, Liu J, Li L, et al. Differential regulation of the REGγ‑proteasome pathway by p53/TGF‑β signalling and mutant p53 in cancer cells. Nat Commun. 2013;4:2667. https://doi.org/10.1038/ncomms3667
123. Matsushita M, Matsuzaki K, Date M, Watanabe T, Shibano K, Nakagawa T, et al. Down‑regulation of TGF‑beta receptors in human colorectal cancer: implications for cancer development. Br J Cancer. 1999;80(1‑2):194‑205. https://doi.org/10.1038/sj.bjc.6690339
124. Lainé A, Labiad O, Hernandez‑Vargas H, This S, Sanlaville A, Léon S, et al. Regulatory T cells promote cancer immune‑escape through integrin αvβ8‑mediated TGF‑β activation. Nat Commun. 2021;12(1):6228. https://doi.org/10.1038/s41467-021-26352-2
125. Qin J, Wu SP, Creighton CJ, Dai F, Xie X, Cheng CM, et al. COUP‑TFII inhibits TGF‑β‑induced growth barrier to promote prostate tumorigenesis. Nature. 2013;493(7431):236‑240. https://doi.org/10.1038/nature11674
126. Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF‑beta in homeostasis and cancer. Nat Rev Cancer. 2003;3(11):807‑821. https://doi.org/10.1038/nrc1208
127. Tauriello DVF, Sancho E, Batlle E. Overcoming TGFβ‑mediated immune evasion in cancer. Nat Rev Cancer. 2022;22(1):25‑44. https://doi.org/10.1038/s41568-021-00413-6
128. Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF‑β‑associated extracellular matrix genes link cancer‑associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun. 2018;9(1):4692. https://doi.org/10.1038/s41467-018-06654-8
129. Li S, Liu M, Do MH, Chou C, Stamatiades EG, Nixon BG, et al. Cancer immunotherapy via targeted TGF‑β signalling blockade in TH cells. Nature. 2020;587(7832):121‑125. https://doi.org/10.1038/s41586-020-2850-3
130. Yingling JM, Blanchard KL, Sawyer JS. Development of TGF‑beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3(12):1011‑1022. https://doi.org/10.1038/nrd1580
131. Visan I. Targeting TGF‑β in cancer. Nat Immunol. 2018;19,316. https://doi.org/10.1038/s41590-018-0076-4
132. Bierie B, Moses HL. Tumour microenvironment: TGF‑beta-the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6(7):506‑520. https://doi.org/10.1038/nrc1926
133. Derynck R, Akhurst RJ, Balmain A. TGF‑beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117‑129. https://doi.org/10.1038/ng1001-117
134. Liu M, Kuo F, Capistrano KJ, Kang D, Nixon BG, Shi W, et al. TGF‑β suppresses type 2 immunity to cancer. Nature. 2020;587(7832):115‑120. https://doi.org/10.1038/s41586-020-2836-1
135. Ahuja N, Ashok C, Natua S, Pant D, Cherian A, Pandkar MR, et al. Hypoxia‑induced TGF‑β‑RBFOX2‑ESRP1 axis regulates human MENA alternative splicing and promotes EMT in breast cancer. NAR Cancer. 2020;2(3):zcaa021. https://doi.org/10.1093/narcan/zcaa021
136. Nasarre P, Gemmill RM, Potiron VA, Roche J, Lu X, Barón AE, et al. Neuropilin‑2 is upregulated in lung cancer cells during TGF‑β1‑induced epithelial‑mesenchymal transition. Cancer Res. 2013;73(23):7111‑7121. https://doi.org/10.1158/0008-5472.can-13-1755
137. Chen M, Wu C, Fu Z, Liu S. ICAM1 promotes bone metastasis via integrin‑mediated TGF‑β/EMT signaling in triple‑negative breast cancer. Cancer Sci. 2022;113(11):3751‑3765. https://doi.org/10.1111/cas.15532.
138. Shen L, Qu X, Ma Y, Zheng J, Chu D, Liu B, et al. Tumor suppressor NDRG2 tips the balance of oncogenic TGF‑β via EMT inhibition in colorectal cancer. Oncogenesis. 2014;3(2):e86. https://doi.org/10.1038/oncsis.2013.48
139. Oktyabri D, Tange S, Terashima M, Ishimura A, Suzuki T. EED regulates epithelial‑mesenchymal transition of cancer cells induced by TGF‑β. Biochem Biophys Res Commun. 2014;453(1):124‑130. https://doi.org/10.1016/j.bbrc.2014.09.082.
140. Sun Z, Su Z, Zhou Z, Wang S, Wang Z, Tong X, et al. RNA demethylase ALKBH5 inhibits TGF‑β‑induced EMT by regulating TGF‑β/SMAD signaling in non‑small cell lung cancer. FASEB J. 2022;36(5):e22283. https://doi.org/10.1096/fj.202200005RR.
141. Pang MF, Georgoudaki AM, Lambut L, Johansson J, Tabor V, Hagikura K, et al. TGF‑β1‑induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21‑mediated chemotaxis. Oncogene. 2016;35(6):748‑760. https://doi.org/10.1038/onc.2015.133
142. Ping Q, Wang C, Cheng X, Zhong Y, Yan R, Yang M, et al. TGF‑β1 dominates stromal fibroblast‑mediated EMT via the FAP/VCAN axis in bladder cancer cells. J Transl Med. 2023;21(1):475. https://doi.org/10.1186/s12967-023-04303-3
143. Fernandes S, Oliver‑De La Cruz J, Morazzo S, Niro F, Cassani M, Duríková H, et al. TGF‑β induces matrisome pathological alterations and EMT in patient‑derived prostate cancer tumoroids. Matrix Biol. 2024;125:12‑30. https://doi.org/10.1016/j.matbio.2023.11.001.
144. Zhang X, Zhang P, Shao M, Zang X, Zhang J, Mao F, et al. SALL4 activates TGF-β/SMAD signaling pathway to induce EMT and promote gastric cancer metastasis. Cancer Manag Res. 2018;10:4459‑4470. https://doi.org/10.2147/CMAR.S177373
145. Zhao X, Wu X, Qian M, Song Y, Wu D, Zhang W. Knockdown of TGF‑β1 expression in human umbilical cord mesenchymal stem cells reverts their exosome‑mediated EMT promoting effect on lung cancer cells. Cancer Lett. 2018;428:34‑44. https://doi.org/10.1016/j.canlet.2018.04.026.
146. Chen XH, Liu ZC, Zhang G, Wei W, Wang XX, Wang H, et al. TGF‑β and EGF induced HLA‑I downregulation is associated with epithelial‑mesenchymal transition (EMT) through upregulation of snail in prostate cancer cells. Mol Immunol. 2015;65(1):34‑42. https://doi.org/10.1016/j.molimm.2014.12.017.
147. Zhang H, Liu L, Wang Y, Zhao G, Xie R, Liu C, et al. KLF8 involves in TGF‑beta‑induced EMT and promotes invasion and migration in gastric cancer cells. J Cancer Res Clin Oncol. 2013;139(6):1033‑1042. https://doi.org/10.1007/s00432-012-1363-3
148. Gao J, Zhu Y, Nilsson M, Sundfeldt K. TGF‑β isoforms induce EMT‑independent migration of ovarian cancer cells. Cancer Cell Int. 2014;14(1):72. https://doi.org/10.1186/s12935-014-0072-1
149. Kim BN, Ahn DH, Kang N, Yeo CD, Kim YK, Lee KY, et al. TGF‑β induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci Rep. 2020;10(1):10597. https://doi.org/10.1038/s41598-020-67325-7
150. Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer‑associated fibroblasts induce epithelial‑mesenchymal transition of breast cancer cells through paracrine TGF‑β signalling. Br J Cancer. 2014;110(3):724‑732. https://doi.org/10.1038/bjc.2013.768
151. Wu YY, Peck K, Chang YL, Pan SH, Cheng YF, Lin JC, et al. SCUBE3 is an endogenous TGF‑β receptor ligand and regulates the epithelial‑mesenchymal transition in lung cancer. Oncogene. 2011;30(34):3682‑3693. https://doi.org/10.1038/onc.2011.85
152. Zhang Z, Xu Y. FZD7 accelerates hepatic metastases in pancreatic cancer by strengthening EMT and stemness associated with TGF‑β/SMAD3 signaling. Mol Med. 2022;28(1):82. https://doi.org/10.1186/s10020-022-00509-1
153. Hirakawa M, Takimoto R, Tamura F, Yoshida M, Ono M, Murase K, et al. Fucosylated TGF‑β receptors transduces a signal for epithelial‑mesenchymal transition in colorectal cancer cells. Br J Cancer. 2014;110(1):156‑163. https://doi.org/10.1038/bjc.2013.699
154. Hiraga R, Kato M, Miyagawa S, Kamata T. Nox4‑derived ROS signaling contributes to TGF‑β‑induced epithelial‑mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2013;33(10):4431‑4438. PMID: 24123012
155. Cardenas H, Vieth E, Lee J, Segar M, Liu Y, Nephew KP, et al. TGF‑β induces global changes in DNA methylation during the epithelial‑to‑mesenchymal transition in ovarian cancer cells. Epigenetics. 2014;9(11):1461‑1472. https://doi.org/10.4161/15592294.2014.971608
156. Ko H, So Y, Jeon H, Jeong MH, Choi HK, Ryu SH, et al. TGF‑β1‑induced epithelial‑mesenchymal transition and acetylation of Smad2 and Smad3 are negatively regulated by EGCG in human A549 lung cancer cells. Cancer Lett. 2013;335(1):205‑213. https://doi.org/10.1016/j.canlet.2013.02.018
157. Araki K, Shimura T, Suzuki H, Tsutsumi S, Wada W, Yajima T, et al. E/N‑cadherin switch mediates cancer progression via TGF‑β‑induced epithelial‑to‑mesenchymal transition in extrahepatic cholangiocarcinoma. Br J Cancer. 2011;105(12):1885‑1893. https://doi.org/10.1038/bjc.2011.452
158. Li Y, Wang P, Ye D, Bai X, Zeng X, Zhao Q, et al. IGHG1 induces EMT in gastric cancer cells by regulating TGF‑β/SMAD3 signaling pathway. J Cancer. 2021;12(12):3458‑3467. https://doi.org/10.7150/jca.56056
159. Sethi N, Kang Y. Notch signalling in cancer progression and bone metastasis. Br J Cancer. 2011;105(12):1805‑1810. https://doi.org/10.1038/bjc.2011.497
160. Roper N, Velez MJ, Chiappori A, Kim YS, Wei JS, Sindiri S, et al. Notch signaling and efficacy of PD‑1/PD‑L1 blockade in relapsed small cell lung cancer. Nat Commun. 2021;12(1):3880. https://doi.org/10.1038/s41467-021-24164-y
161. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338‑351. https://doi.org/10.1038/nrc3035
162. Dotto GP. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat Rev Cancer. 2009;9(8):587‑595. https://doi.org/10.1038/nrc2675
163. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8(2):97‑106. https://doi.org/10.1038/nrclinonc.2010.196
164. Majumder S, Crabtree JS, Golde TE, Minter LM, Osborne BA, Miele L. Targeting Notch in oncology: the path forward. Nat Rev Drug Discov. 2021;20(2):125‑144. https://doi.org/10.1038/s41573-020-00091-3
165. Lim JS, Ibaseta A, Fischer MM, Cancilla B, O’Young G, Cristea S, et al. Intratumoural heterogeneity generated by Notch signalling promotes small‑cell lung cancer. Nature. 2017;545(7654):360‑364. https://doi.org/10.1038/nature22323
166. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12(8):445‑464. https://doi.org/10.1038/nrclinonc.2015.61
167. Rampias T, Vgenopoulou P, Avgeris M, Polyzos A, Stravodimos K, Valavanis C, et al. A new tumor suppressor role for the Notch pathway in bladder cancer. Nat Med. 2014;20(10):1199‑1205. https://doi.org/10.1038/nm.3678
168. Maraver A, Fernandez‑Marcos PJ, Cash TP, Mendez‑Pertuz M, Duenas M, Maietta P, et al. NOTCH pathway inactivation promotes bladder cancer progression. J Clin Invest. 2015;125(2):824‑830. https://doi.org/10.1172/JCI78185
169. Fender AW, Nutter JM, Fitzgerald TL, Bertrand FE, Sigounas G. Notch‑1 promotes stemness and epithelial to mesenchymal transition in colorectal cancer. J Cell Biochem. 2015;116(11):2517‑2527. https://doi.org/10.1002/jcb.25196
170. Zang M, Hu L, Fan ZY, Wang HX, Zhu ZL, Cao S, et al. Luteolin suppresses gastric cancer progression by reversing epithelial‑mesenchymal transition via suppression of the Notch signaling pathway. J Transl Med. 2017;15(1):52. https://doi.org/10.1186/s12967-017-1151-6
171. Bocci F, Jolly MK, Tripathi SC, Aguilar M, Hanash SM, Levine H, Onuchic JN. Numb prevents a complete epithelial‑mesenchymal transition by modulating Notch signalling. J R Soc Interface. 2017;14(136):20170512. https://doi.org/10.1098/rsif.2017.0512
172. Zhang L, Sha J, Yang G, Huang X, Bo J, Huang Y. Activation of Notch pathway is linked with epithelial‑mesenchymal transition in prostate cancer cells. Cell Cycle. 2017;16(10):999‑1007. https://doi.org/10.1080/15384101.2017.1312237
173. Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, et al. Acquisition of epithelial‑mesenchymal transition phenotype of gemcitabine‑resistant pancreatic cancer cells is linked with activation of the Notch signaling pathway. Cancer Res. 2009;69(6):2400‑2407. https://doi.org/10.1158/0008-5472.can-08-4312
174. Zhang J, Kuang Y, Wang Y, Xu Q, Ren Q. Notch‑4 silencing inhibits prostate cancer growth and EMT via the NF‑κB pathway. Apoptosis. 2017;22(6):877‑884. https://doi.org/10.1007/s10495-017-1368-0
175. Zhou J, Jain S, Azad AK, Xu X, Yu HC, Xu Z, et al. Notch and TGFβ form a positive regulatory loop and regulate EMT in epithelial ovarian cancer cells. Cell Signal. 2016;28(8):838‑849. https://doi.org/10.1016/j.cellsig.2016.03.016.
176. Keysar SB, Le PN, Anderson RT, Morton JJ, Bowles DW, Paylor JJ, et al. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti‑EGFR therapeutic resistance in head and neck cancer. Cancer Res. 2013;73(11):3381‑3392. https://doi.org/10.1158/0008-5472.can-12-4047
177. Yue D, Li H, Che J, Zhang Y, Tseng HHK, Jin JQ, et al. Hedgehog/Gli promotes epithelial‑mesenchymal transition in lung squamous cell carcinomas. J Exp Clin Cancer Res. 2014;33(1):34. https://doi.org/10.1186/1756-9966-33-34
178. Zhang K, Sun C, Zhang Q, Wang X. Sonic hedgehog‑Gli1 signals promote epithelial‑mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Med Oncol. 2015;32(1):368. https://doi.org/10.1007/s12032-014-0368-y
179. Xu X, Zhou Y, Xie C, Wei S, Gan H, He S, et al. Genome‑wide screening reveals an EMT molecular network mediated by Sonic hedgehog‑Gli1 signaling in pancreatic cancer cells. PLoS One. 2012;7(8):e43119. https://doi.org/10.1371/journal.pone.0043119
180. Sun M, Zhang N, Wang X, Li Y, Qi W, Zhang H, et al. Hedgehog pathway is involved in nitidine chloride induced inhibition of epithelial‑mesenchymal transition and cancer stem cell‑like properties in breast cancer cells. Cell Biosci. 2016;6:44. https://doi.org/10.1186/s13578-016-0104-8
181. Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q, et al. Hedgehog signaling regulates hypoxia‑induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand‑independent manner. Mol Cancer. 2013;12:66. https://doi.org/10.1186/1476-4598-12-66
182. Wang F, Ma L, Zhang Z, Liu X, Gao H, Zhuang Y, et al. Hedgehog signaling regulates epithelial‑mesenchymal transition in pancreatic cancer stem‑like cells. J Cancer. 2016;7(4):408‑417. https://doi.org/10.7150/jca.13305
183. Yoo YA, Kang MH, Lee HJ, Kim B, Park JK, Kim HK, et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP‑9 pathway in gastric cancer. Cancer Res. 2011;71(22):7061‑7070. https://doi.org/10.1158/0008-5472.can-11-1338
184. Ahmad A, Maitah MY, Ginnebaugh KR, Li Y, Bao B, Gadgeel SM, et al. Inhibition of Hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT‑regulating miRNAs. J Hematol Oncol. 2013;6(1):77. https://doi.org/10.1186/1756-8722-6-77
185. Xu X, Su B, Xie C, Wei S, Zhou Y, Liu H, et al. Sonic hedgehog‑Gli1 signaling pathway regulates the epithelial‑mesenchymal transition (EMT) by mediating a new target gene, S100A4, in pancreatic cancer cells. PLoS One. 2014 Jul 29;9(7):e96441. https://doi.org/10.1371/journal.pone.0096441
186. Islam SS, Mokhtari RB, Noman AS, Uddin M, Rahman MZ, Azadi MA, et al. Sonic hedgehog (Shh) signaling promotes tumorigenicity and stemness via activation of epithelial‑to‑mesenchymal transition (EMT) in bladder cancer. Mol Carcinog. 2016;55(5):537‑551. https://doi.org/10.1002/mc.22300
187. Yamamichi F, Shigemura K, Behnsawy HM, Meligy FY, Huang WC, Li X, et al. Sonic hedgehog and androgen signaling in tumor and stromal compartments drives epithelial‑mesenchymal transition in prostate cancer. Scand J Urol. 2014;48(6):523‑532. https://doi.org/10.3109/21681805.2014.898336
188. Batsaikhan BE, Yoshikawa K, Kurita N, Iwata T, Takasu C, Kashihara H, et al. Cyclopamine decreased the expression of Sonic Hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res. 2014 Nov;34(11):6339-44. PMID: 25368233
189. Ke B, Wang XN, Liu N, Li B, Wang XJ, Zhang RP, et al. Sonic Hedgehog/Gli1 signaling pathway regulates cell migration and invasion via induction of epithelial‑to‑mesenchymal transition in gastric cancer. J Cancer. 2020;11(13):3932‑3943. https://doi.org/10.7150/jca.42900
190. Cao L, Xiao X, Lei J, Duan W, Ma Q, Li W. Curcumin inhibits hypoxia‑induced epithelial‑mesenchymal transition in pancreatic cancer cells via suppression of the hedgehog signaling pathway. Oncol Rep. 2016;35(6):3728‑3734. https://doi.org/10.3892/or.2016.4709
191. Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W, et al. SDF‑1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial‑mesenchymal transition in vitro through non‑canonical activation of Hedgehog pathway. Cancer Lett. 2012;322(2):169‑176. https://doi.org/10.1016/j.canlet.2012.02.035.
192. Huber MA, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, et al. NF‑kappaB is essential for epithelial‑mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114(4):569‑581. https://doi.org/10.1172/JCI21358
193. Pires BRB, Mencalha AL, Ferreira GM, de Souza WF, Morgado‑Díaz JA, Maia AM, et al. NF‑kappaB is involved in the regulation of EMT genes in breast cancer cells. PLoS One. 2017;12(1):e0169622. https://doi.org/10.1371/journal.pone.0169622
194. Zhou P, Wang C, Hu Z, Chen W, Qi W, Li A. Genistein induces apoptosis of colon cancer cells by reversal of epithelial‑to‑mesenchymal transition via a Notch1/NF‑κB/Slug/E‑cadherin pathway. BMC Cancer. 2017;17(1):813. https://doi.org/10.1186/s12885-017-3829-9
195. Luo M, Hou L, Li J, Shao S, Huang S, Meng D, et al. VEGF/NRP‑1 axis promotes progression of breast cancer via enhancement of epithelial‑mesenchymal transition and activation of NF‑κB and β‑catenin. Cancer Lett. 2016;373(1):1‑11. https://doi.org/10.1016/j.canlet.2016.01.010.
196. Li QQ, Chen ZQ, Cao XX, Xu JD, Xu JW, Chen YY, et al. Involvement of NF‑κB/miR‑448 regulatory feedback loop in chemotherapy‑induced epithelial‑mesenchymal transition of breast cancer cells. Cell Death Differ. 2011;18(1):16‑25. https://doi.org/10.1038/cdd.2010.103
197. Maier HJ, Schmidt‑Strassburger U, Huber MA, Wiedemann EM, Beug H, Wirth T. NF‑kappaB promotes epithelial‑mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010;295(2):214‑228. https://doi.org/10.1016/j.canlet.2010.03.003.
198. Cichon MA, Radisky DC. ROS‑induced epithelial‑mesenchymal transition in mammary epithelial cells is mediated by NF‑κB‑dependent activation of Snail. Oncotarget. 2014;5(9):2827‑2838. https://doi.org/10.18632/oncotarget.1940
199. Nomura A, Majumder K, Giri B, Dauer P, Dudeja V, Roy S, et al. Inhibition of NF‑kappa B pathway leads to deregulation of epithelial‑mesenchymal transition and neural invasion in pancreatic cancer. Lab Invest. 2016;96(12):1268‑1278. https://doi.org/10.1038/labinvest.2016.109
200. Zhang L, Wang D, Li Y, Liu Y, Xie X, Wu Y, et al. CCL21/CCR7 axis contributed to CD133+ pancreatic cancer stem‑like cell metastasis via EMT and Erk/NF‑κB pathway. PLoS One. 2016;11(8):e0158529. https://doi.org/10.1371/journal.pone.0158529
201. Chua HL, Bhat‑Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF‑kappaB represses E‑cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB‑1 and ZEB‑2. Oncogene. 2007;26(5):711‑724. https://doi.org/10.1038/sj.onc.1209808
202. Wang J, Tian L, Khan MN, Zhang L, Chen Q, Zhao Y, et al. Ginsenoside Rg3 sensitizes hypoxic lung cancer cells to cisplatin via blocking of NF‑κB‑mediated epithelial‑mesenchymal transition and stemness. Cancer Lett. 2018;415:73‑85. https://doi.org/10.1016/j.canlet.2017.11.037.
203. Esparza‑López J, Alvarado‑Munoz JF, Escobar‑Arriaga E, Ulloa‑Aguirre A, Ibarra‑Sánchez MJ. Metformin reverses mesenchymal phenotype of primary breast cancer cells through STAT3/NF‑κB pathways. BMC Cancer. 2019;19(1):728. https://doi.org/10.1186/s12885-019-5945-1
204. Rigillo G, Belluti S, Campani V, Ragazzini G, Ronzio M, Miserocchi G, et al. The NF‑Y splicing signature controls hybrid EMT and ECM‑related pathways to promote aggressiveness of colon cancer. Cancer Lett. 2023;567:216262. https://doi.org/10.1016/j.canlet.2023.216262.
205. Maeda K, Ding Q, Yoshimitsu M, Kuwahata T, Miyazaki Y, Tsukasa K, et al. CD133 modulates HIF‑1α expression under hypoxia in EMT phenotype pancreatic cancer stem‑like cells. Int J Mol Sci. 2016;17(7):1025. https://doi.org/10.3390/ijms17071025
206. Cheng ZX, Sun B, Wang SJ, Gao Y, Zhang YM, Zhou HX, et al. Nuclear factor‑κB‑dependent epithelial to mesenchymal transition induced by HIF‑1α activation in pancreatic cancer cells under hypoxic conditions. PLoS One. 2011;6(8):e23752. https://doi.org/10.1371/journal.pone.0023752
207. Huang YN, Xu YY, Ma Q, Li MQ, Guo JX, Wang X, et al. Dextran sulfate effects EMT of human gastric cancer cells by reducing HIF‑1α/TGF‑β. J Cancer. 2021;12(11):3367‑3377. https://doi.org/10.7150/jca.55550
208. Zhao JH, Luo Y, Jiang YG, He DL, Wu CT. Knockdown of β‑catenin through shRNA causes a reversal of EMT and metastatic phenotypes induced by HIF‑1α. Cancer Invest. 2011;29(6):377‑382. https://doi.org/10.3109/07357907.2010.512595
209. Zhang W, Shi X, Peng Y, Wu M, Zhang P, Xie R, et al. HIF‑1α promotes epithelial‑mesenchymal transition and metastasis through direct regulation of ZEB1 in colorectal cancer. PLoS One. 2015;10(6):e0129603. https://doi.org/10.1371/journal.pone.0129603
210. Liu T, Zhao L, Zhang Y, Chen W, Liu D, Hou H, et al. Ginsenoside 20(S)‑Rg3 targets HIF‑1α to block hypoxia‑induced epithelial‑mesenchymal transition in ovarian cancer cells. PLoS One. 2014;9(9):e103887. https://doi.org/10.1371/journal.pone.0103887
211. Luo F, Lu FT, Cao JX, Ma WJ, Xia ZF, Zhan JH, et al. HIF‑1α inhibition promotes the efficacy of immune checkpoint blockade in the treatment of non‑small cell lung cancer. Cancer Lett. 2022;531:39‑56. https://doi.org/10.1016/j.canlet.2022.01.027.
212. Wang M, Yan J, Cao X, Hua P, Li Z. Hydrogen sulfide modulates epithelial‑mesenchymal transition and angiogenesis in non‑small cell lung cancer via HIF‑1α activation. Biochem Pharmacol. 2020;172:113775. https://doi.org/10.1016/j.bcp.2019.113775.
213. Luo Y, Lan L, Jiang YG, Zhao JH, Li MC, Wei NB, et al. Epithelial‑mesenchymal transition and migration of prostate cancer stem cells is driven by cancer‑associated fibroblasts in an HIF‑1α/β‑catenin‑dependent pathway. Mol Cells. 2013;36(2):138‑144. https://doi.org/10.1007/s10059-013-0096-8
214. Zhang J, Zhu L, Fang J, Ge Z, Li X. LRG1 modulates epithelial‑mesenchymal transition and angiogenesis in colorectal cancer via HIF‑1α activation. J Exp Clin Cancer Res. 2016;35:29. https://doi.org/10.1186/s13046-016-0306-2
215. Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, Liu J, Wang Q, Zhu J, Feng X, Dong J, Qian C. Hypoxia induces epithelial‑mesenchymal transition via activation of SNAI1 by hypoxia‑inducible factor‑1α in hepatocellular carcinoma. BMC Cancer. 2013;13:108. https://doi.org/10.1186/1471-2407-13-108
216. Wang M, Zhao X, Zhu D, Liu T, Liang X, Liu F, et al. HIF‑1α promoted vasculogenic mimicry formation in hepatocellular carcinoma through LOXL2 up‑regulation in hypoxic tumor microenvironment. J Exp Clin Cancer Res. 2017;36(1):60. https://doi.org/10.1186/s13046-017-0533-1
217. Yoo YG, Christensen J, Gu J, Huang LE. HIF‑1α mediates tumor hypoxia to confer a perpetual mesenchymal phenotype for malignant progression. Sci Signal. 2011 Jun 21;4(178):pt4. https://doi.org/10.1126/scisignal.2002072
218. Yeh YH, Hsiao HF, Yeh YC, Chen TW, Li TK. Inflammatory interferon activates HIF‑1α‑mediated epithelial‑to‑mesenchymal transition via PI3K/AKT/mTOR pathway. J Exp Clin Cancer Res. 2018;37(1):70. https://doi.org/10.1186/s13046-018-0730-6
219. Bocci F, Tripathi SC, Vilchez Mercedes SA, George JT, Casabar JP, Wong PK, et al. NRF2 activates a partial epithelial‑mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr Biol (Camb). 2019;11(6):251‑263. https://doi.org/10.1093/intbio/zyz021
220. Feng R, Morine Y, Ikemoto T, Imura S, Iwahashi S, Saito Y, et al. Nrf2 activation drives macrophage polarization and cancer cell epithelial‑mesenchymal transition during interaction. Cell Commun Signal. 2018;16(1):54. https://doi.org/10.1186/s12964-018-0262-x
221. Yazaki K, Matsuno Y, Yoshida K, Sherpa M, Nakajima M, Matsuyama M, et al. ROS‑Nrf2 pathway mediates the development of TGF‑β1‑induced epithelial‑mesenchymal transition through the activation of Notch signaling. Eur J Cell Biol. 2021;100(7‑8):151181. https://doi.org/10.1016/j.ejcb.2021.151181.
222. Dong P, Xiong Y, Watari H, Hanley SJB, Konno Y, Ihira K, et al. MiR‑137 and miR‑34a directly target Snail and inhibit EMT, invasion and sphere‑forming ability of ovarian cancer cells. J Exp Clin Cancer Res. 2016;35(1):132. https://doi.org/10.1186/s13046-016-0415-y
223. You J, Li Y, Fang N, Liu B, Zu L, Chang R, et al. MiR‑132 suppresses the migration and invasion of lung cancer cells via targeting the EMT regulator ZEB2. PLoS One. 2014;9(3):e91827. https://doi.org/10.1371/journal.pone.0091827
224. Sun L, Yao Y, Liu B, Lin Z, Lin L, Yang M, et al. MiR‑200b and miR‑15b regulate chemotherapy‑induced epithelial‑mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene. 2012;31(4):432‑445. https://doi.org/10.1038/onc.2011.263
225. Harazono Y, Muramatsu T, Endo H, Uzawa N, Kawano T, Harada K, et al. miR‑655 is an EMT‑suppressive microRNA targeting ZEB1 and TGFBR2. PLoS One. 2013;8(5):e62757. https://doi.org/10.1371/journal.pone.0062757
226. Liu YN, Yin JJ, Abou‑Kheir W, Hynes PG, Casey OM, Fang L, et al. MiR‑1 and miR‑200 inhibit EMT via Slug‑dependent and tumorigenesis via Slug‑independent mechanisms. Oncogene. 2013 Jan 17;32(3):296‑306. https://doi.org/10.1038/onc.2012.58
227. Arora H, Qureshi R, Park WY. miR‑506 regulates epithelial mesenchymal transition in breast cancer cell lines. PLoS One. 2013;8(5):e64273. https://doi.org/10.1371/journal.pone.0064273
228. Zhu Y, Huang C, Zhang C, Zhou Y, Zhao E, Zhang Y, et al. LncRNA MIR200CHG inhibits EMT in gastric cancer by stabilizing miR‑200c from target‑directed miRNA degradation. Nat Commun. 2023;14(1):8141. https://doi.org/10.1038/s41467-023-43974-w
229. Zhou W, Ye XL, Xu J, Cao MG, Fang ZY, Li LY, Guan GH, Liu Q, Qian YH, Xie D. The lncRNA H19 mediates breast cancer cell plasticity during EMT and MET plasticity by differentially sponging miR‑200b/c and let‑7b. Sci Signal. 2017;10(483):eaak9557. https://doi.org/10.1126/scisignal.aak9557
230. Xu L, Liu W, Li T, Hu Y, Wang Y, Huang L, et al. Long non‑coding RNA SMASR inhibits the EMT by negatively regulating TGF‑β/Smad signaling pathway in lung cancer. Oncogene. 2021;40(20):3578‑3592. https://doi.org/10.1038/s41388-021-01760-2
231. Meng X, Xiao W, Sun J, Li W, Yuan H, Yu T, et al. CircPTK2/PABPC1/SETDB1 axis promotes EMT‑mediated tumor metastasis and gemcitabine resistance in bladder cancer. Cancer Lett. 2023;554:216023. https://doi.org/10.1016/j.canlet.2022.216023.
232. Yu Z, Zhu X, Li Y, Liang M, Liu M, Liu Z, et al. Circ‑HMGA2 (hsa_circ_0027446) promotes the metastasis and epithelial‑mesenchymal transition of lung adenocarcinoma cells through the miR‑1236‑3p/ZEB1 axis. Cell Death Dis. 2021;12(4):313. https://doi.org/10.1038/s41419-021-03601-2
233. Imodoye SO, Adedokun KA. EMT‑induced immune evasion: connecting the dots from mechanisms to therapy. Clin Exp Med. 2023;23(8):4265‑4287. https://doi.org/10.1007/s10238-023-01229-4
234. Hu B, Tian X, Li Y, Liu Y, Yang T, Han Z, et al. Epithelial‑mesenchymal transition may be involved in the immune evasion of circulating gastric tumor cells via downregulation of ULBP1. Cancer Med. 2020;9(8):2686‑2697. https://doi.org/10.1002/cam4.2871.
235. Kolijn K, Verhoef EI, Smid M, Böttcher R, Jenster GW, Debets R, et al. Epithelial‑mesenchymal transition in human prostate cancer demonstrates enhanced immune evasion marked by IDO1 expression. Cancer Res. 2018;78(16):4671‑4679. https://doi.org/10.1158/0008-5472.can-17-3752
236. Liang Z, Lu L, Mao J, Li X, Qian H, Xu W. Curcumin reversed chronic tobacco smoke exposure‑induced urocystic EMT and acquisition of cancer stem cell properties via Wnt/β‑catenin. Cell Death Dis. 2017;8(10):e3066. https://doi.org/10.1038/cddis.2017.452
237. Lin CH, Shen YA, Hung PH, Yu YB, Chen YJ. Epigallocathechin gallate, polyphenol present in green tea, inhibits stem‑like characteristics and epithelial‑mesenchymal transition in nasopharyngeal cancer cell lines. BMC Complement Altern Med. 2012;12:201. https://doi.org/10.1186/1472-6882-12-201
238. Wei R, Cortez Penso NE, Hackman RM, Wang Y, Mackenzie GG. Epigallocatechin‑3‑Gallate (EGCG) suppresses pancreatic cancer cell growth, invasion, and migration partly through the inhibition of Akt pathway and epithelial‑mesenchymal transition: enhanced efficacy when combined with gemcitabine. Nutrients. 2019;11(8):1856. https://doi.org/10.3390/nu11081856
239. Tang SN, Singh C, Nall D, Meeker D, Shankar S, Srivastava RK. The dietary bioflavonoid quercetin synergizes with epigallocathechin gallate (EGCG) to inhibit prostate cancer stem cell characteristics, invasion, migration and epithelial‑mesenchymal transition. J Mol Signal. 2010;5:14. https://doi.org/10.1186/1750-2187-5-14
240. Chen PN, Chu SC, Kuo WH, Chou MY, Lin JK, Hsieh YS. Epigallocatechin‑3‑gallate inhibits invasion, epithelial‑mesenchymal transition, and tumor growth in oral cancer cells. J Agric Food Chem. 2011;59(8):3836‑3844. https://doi.org/10.1021/jf1049408
241. Lee GA, Choi KC, Hwang KA. Kaempferol, a phytoestrogen, suppressed triclosan‑induced epithelial‑mesenchymal transition and metastatic‑related behaviors of MCF‑7 breast cancer cells. Environ Toxicol Pharmacol. 2017;49:48‑57. https://doi.org/10.1016/j.etap.2016.11.016
242. Jo E, Park SJ, Choi YS, Jeon WK, Kim BC. Kaempferol suppresses transforming growth factor‑β1‑induced epithelial‑to‑mesenchymal transition and migration of A549 lung cancer cells by inhibiting Akt1‑mediated phosphorylation of Smad3 at threonine‑179. Neoplasia. 2015;17(7):525‑537. https://doi.org/10.1016/j.neo.2015.06.004
243. Chang JH, Lai SL, Chen WS, Hung WY, Chow JM, Hsiao M, et al. Quercetin suppresses the metastatic ability of lung cancer through inhibiting Snail‑dependent Akt activation and Snail‑independent ADAM9 expression pathways. Biochim Biophys Acta Mol Cell Res. 2017;1864(10):1746‑1758. https://doi.org/10.1016/j.bbamcr.2017.06.017
244. Feng J, Song D, Jiang S, Yang X, Ding T, Zhang H, et al. Quercetin restrains TGF‑β1‑induced epithelial‑mesenchymal transition by inhibiting Twist1 and regulating E‑cadherin expression. Biochem Biophys Res Commun. 2018;498(1):132‑138. https://doi.org/10.1016/j.bbrc.2018.02.044
245. Lu X, Chen D, Yang F, Xing N. Quercetin inhibits epithelial‑to‑mesenchymal transition (EMT) process and promotes apoptosis in prostate cancer via downregulating lncRNA MALAT1. Cancer Manag Res. 2020;12:1741‑1750. https://doi.org/10.2147/CMAR.S241093
246. Baribeau S, Chaudhry P, Parent S, Asselin É. Resveratrol inhibits cisplatin‑induced epithelial‑to‑mesenchymal transition in ovarian cancer cell lines. PLoS One. 2014;9(1):e86987. https://doi.org/10.1371/journal.pone.0086987
247. Vergara D, Valente CM, Tinelli A, Siciliano C, Lorusso V, Acierno R, et al. Resveratrol inhibits the epidermal growth factor‑induced epithelial‑mesenchymal transition in MCF‑7 cells. Cancer Lett. 2011;310(1):1‑8. https://doi.org/10.1016/j.canlet.2011.04.009.
248. Dian L, Xu Z, Sun Y, Li J, Lu H, Zheng M, et al. Berberine alkaloids inhibit the proliferation and metastasis of breast carcinoma cells involving Wnt/β‑catenin signaling and EMT. Phytochemistry. 2022;200:113217. https://doi.org/10.1016/j.phytochem.2022.113217
249. Dai W, Wang F, He L, Lin C, Wu S, Chen P, et al. Genistein inhibits hepatocellular carcinoma cell migration by reversing the epithelial‑mesenchymal transition: partial mediation by the transcription factor NFAT1. Mol Carcinog. 2015;54(4):301‑311. https://doi.org/10.1002/mc.22100
250. Kim YS, Choi KC, Hwang KA. Genistein suppressed epithelial‑mesenchymal transition and migration efficacies of BG‑1 ovarian cancer cells activated by estrogenic chemicals via estrogen receptor pathway and downregulation of TGF‑β signaling pathway. Phytomedicine. 2015;22(11):993‑999. https://doi.org/10.1016/j.phymed.2015.08.003
251. Li YW, Xu J, Zhu GY, Huang ZJ, Lu Y, Li XQ, Wang N, Zhang FX. Apigenin suppresses the stem cell‑like properties of triple‑negative breast cancer cells by inhibiting YAP/TAZ activity. Cell Death Discov. 2018;4:105. https://doi.org/10.1038/s41420-018-0124-8
252. Chien MH, Lin YW, Wen YC, Yang YC, Hsiao M, Chang JL, et al. Targeting the SPOCK1‑snail/slug axis‑mediated epithelial‑to‑mesenchymal transition by apigenin contributes to repression of prostate cancer metastasis. J Exp Clin Cancer Res. 2019;38(1):246. https://doi.org/10.1186/s13046-019-1247-3
253. Wang DX, Zou YJ, Zhuang XB, Chen SX, Lin Y, Li WL, et al. Sulforaphane suppresses EMT and metastasis in human lung cancer through miR‑616‑5p‑mediated GSK3β/β‑catenin signaling pathways. Acta Pharmacol Sin. 2017;38(2):241‑251. https://doi.org/10.1038/aps.2016.122
254. Zou T, Lan M, Liu F, Li L, Cai T, Tian H, Cai Y. Emodin‑loaded polymer‑lipid hybrid nanoparticles enhance the sensitivity of breast cancer to doxorubicin by inhibiting epithelial-mesenchymal transition. Cancer Nanotechnol. 2021;12:22. https://doi.org/10.1186/s12645-021-00093-9
255. Shahin SA, Wang R, Simargi SI, Contreras A, Parra Echavarria L, Qu L, et al. Hyaluronic acid conjugated nanoparticle delivery of siRNA against TWIST reduces tumor burden and enhances sensitivity to cisplatin in ovarian cancer. Nanomedicine. 2018;14(4):1381‑1394. https://doi.org/10.1016/j.nano.2018.04.008
256. Gao Y, Zhu J, Zheng X, Lin G, Miao P. Luteolin functionalized zinc oxide nanoparticles for cancer therapy based on autophagy activation and EMT inhibition. Langmuir. 2024;40(49):26363‑26369. https://doi.org/10.1021/acs.langmuir.4c04194
257. Sureban SM, May R, Mondalek FG, Qu D, Ponnurangam S, Pantazis P, et al. Nanoparticle‑based delivery of siDCAMKL‑1 increases microRNA‑144 and inhibits colorectal cancer tumor growth via a Notch‑1 dependent mechanism. J Nanobiotechnology. 2011;9:40. https://doi.org/10.1186/1477-3155-9-40
258. Watson KD, Lai CY, Qin S, Kruse DE, Lin YC, Seo JW, et al. Ultrasound increases nanoparticle delivery by reducing intratumoral pressure and increasing transport in epithelial and epithelial‑mesenchymal transition tumors. Cancer Res. 2012;72(6):1485‑1493. https://doi.org/10.1158/0008-5472.can-11-3232
259. Parvani JG, Gujrati MD, Mack MA, Schiemann WP, Lu ZR. Silencing β3 integrin by targeted ECO/siRNA nanoparticles inhibits EMT and metastasis of triple‑negative breast cancer. Cancer Res. 2015;75(11):2316‑2325. https://doi.org/10.1158/0008-5472.can-14-3485
Declarations
Funding Statement: This work did not receive financial support.
Declaration of Competing Interest: The authors declare no financial or personal relationships with other individuals or organizations that could inappropriately influence or bias the content of this work. All authors have read the final version of the manuscript and confirm that there are no competing interests.
Consent for publication: All authors have approved the final version of the manuscript.
Use of Artificial Intelligence (AI) Disclosure: This article was written by human contributors. Artificial intelligence-based tools were used to improve grammar, language, and readability, without affecting the scientific content, data interpretation, or conclusions of the article. The authors reviewed and verified all content to ensure its accuracy and integrity.
Dual Use Research of Concern (DURC): “The authors confirm that this research does not constitute DURC as defined by the U.S. Government Policy for Oversight of Life Sciences DURC.”
Data Availability Statements (DAS): “No datasets were generated or analyzed in the current study.”
Authors’ affiliations
1- Science Research Center, the Huizhou Central People’s Hospital, Guangdong Medical University, Huizhou, Guangdong, China
2- Independent Researcher, Victoria, British Columbia, V8 V 1P7, Canada
3- Department of Radiation Oncology, Shandong Provincial Key Laboratory of Radiation Oncology, Shandong Cancer Hospital and Institute, Shandong First Medical University, Shandong Academy of Medical Sciences, Jinan, 250117, Shandong, China
# Correspondence: Milad Ashrafizadeh Email: dvm.milad1994@gmail.com
CRediT authorship contribution statement: YT, Writing-original draft, Writing-review editing; NN, Writing-review editing; MA, Conceptualization, Writing-review editing, Writing-original draft, Supervision, Software. All authors contributed to the work and approved the final version of the manuscript.
ORCID ID:Milad Ashrafizadeh: https://orcid.org/0000-0001-6605-822X
Noushin Nabavi: https://orcid.org/0000-0002-5471-6229
Yu Tian: https://orcid.org/0009-0005-0614-9572
How to cite
Tian Y, Nabavi N, Ashrafizadeh M. Epithelial-Mesenchymal Transition in Cancer. Cancer Biome and Targeted Therapy. Published online 2025 Dec.