REVIEW ARTICLE OPEN ACCESS
Tumor Microenvironment in Triple-Negative Breast Cancer and Targeting Approaches
Youli Li1*, Yang Chen2,3*, Yuxin Mu2,3*, Xuemei Xiu3,4*, Wenxing Qin1,2,3#
Received 2025 Aug 10
Accepted 2025 Oct 12
Epub ahead of print: December 2025
Published in issue 2026 Feb 15
Correspondence: Wenxing Qin - Email: qinwenxingqwx@163.com
The author’s information is available at the end of the article.
© 2026 The Author(s). Published by GCINC Press. Open Access licensed under a Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author(s) and source are credited. To view a copy: https://creativecommons.org/licenses/by/4.0/
Abstract
Triple-negative breast cancer (TNBC) is an aggressive and heterogeneous subtype of breast cancer with high recurrence and early metastasis. Unlike hormone receptor-positive or HER2-positive cancers, TNBC lacks targeted therapies, and standard chemotherapy often yields limited and transient responses, making treatment challenging. The tumor microenvironment (TME) plays a central role in TNBC progression, immune evasion, and therapy resistance. It comprises multiple cellular components, tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), tumor-infiltrating lymphocytes (TILs), and myeloid-derived suppressor cells (MDSCs), as well as structural and signaling elements such as the extracellular matrix (ECM), growth factors, and cytokines. Interactions among these components create an immunosuppressive, pro-tumorigenic milieu that supports cancer cell survival, invasion, and metastasis. Targeting the TME has emerged as a promising therapeutic strategy. Immunotherapies, particularly immune checkpoint inhibitors (ICIs), can restore antitumor immunity by reversing T cell exhaustion and mitigating immune suppression. Response rates remain variable, leading to the exploration of combination approaches that pair ICIs with chemotherapy, radiotherapy, or TME-modulating agents to enhance efficacy. Direct targeting of TME components, including CAFs, TAMs, MDSCs, and ECM remodeling enzymes, is also being developed to disrupt the supportive tumor niche and enhance drug delivery. This review provides a comprehensive overview of the TNBC TME, emphasizing its role in tumor progression and therapy resistance, and summarizes current and emerging strategies to target the TME. By clarifying complex cellular and molecular interactions, these approaches aim to sensitize tumors to therapy, prevent metastasis, and support the development of more effective, personalized treatments for TNBC.
Keywords: Tumor microenvironment, triple-negative breast cancer, targeting therapy, immune checkpoint inhibitors, drug resistance, biomarkers.
1. Introduction
Global breast cancer incidence rates are increasing, accounting for 31% of female cancers, and the disease burden is projected to rise by 40% by 2040 (1, 2). Triple-negative breast cancer (TNBC) lacks estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) expression (3, 4). TNBC is the most aggressive breast cancer subtype, representing 10-15% of all breast cancer cases globally (5-8). TNBC is classified into four molecular subtypes under the TNBCtype-4 scheme: basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal (M), and luminal androgen receptor (LAR) (9). These subtypes have distinct characteristics and different responses to therapy and prognosis (9-11) (Figure 1). Approximately 25% of TNBC patients have germline BRCA1/2 mutations (12). The absence of conventional therapeutic targets renders TNBC difficult to manage, with treatment primarily dependent on traditional chemotherapy that often yields limited efficacy. Consequently, TNBC is associated with high recurrence and metastasis rates, resulting in poor patient prognosis (13-18). For advanced-stage TNBC, the median survival remains less than 24 months (19-22).
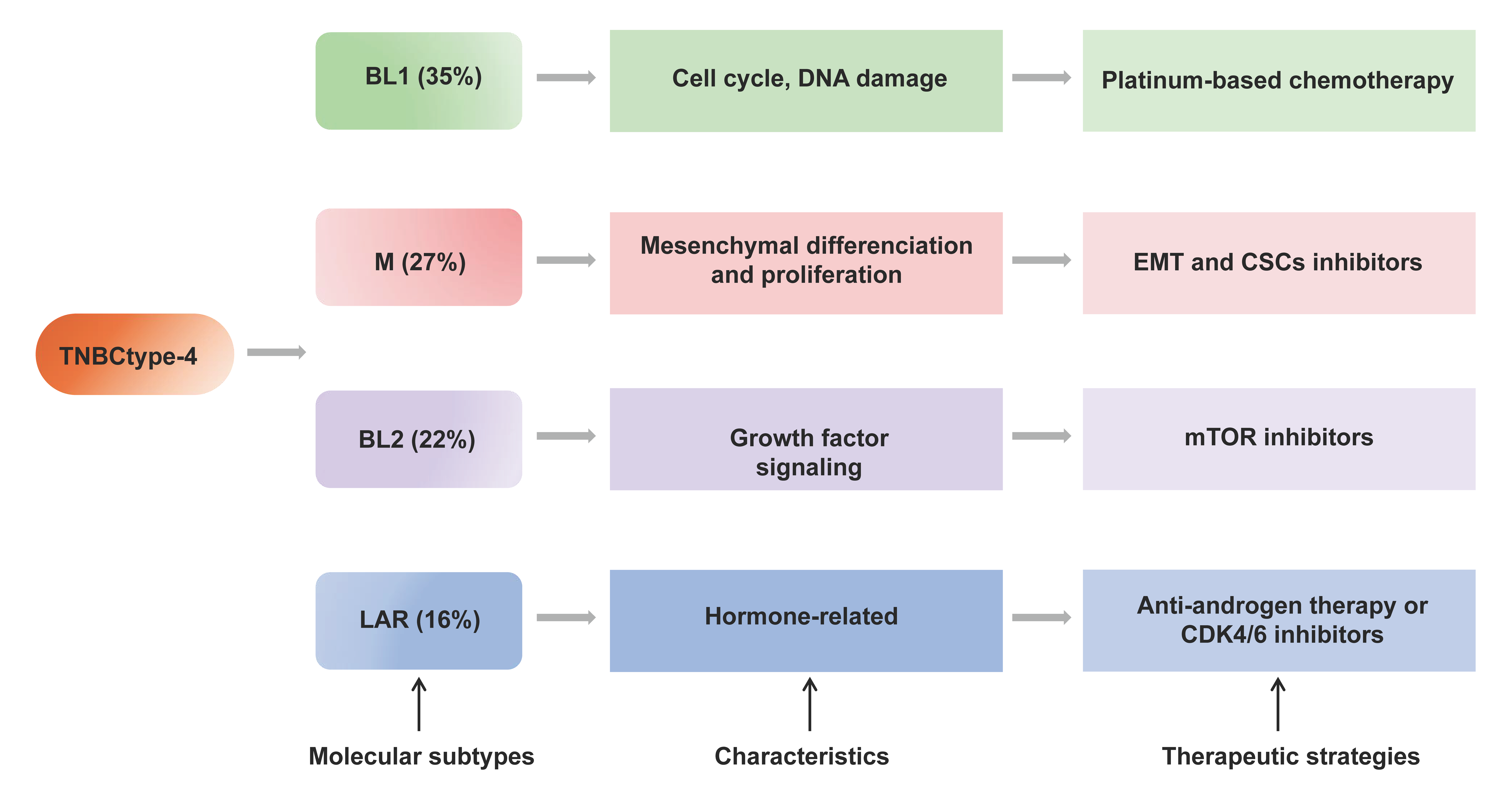
Figure 1: Molecular subtypes, characteristics, and potential therapeutic strategies of TNBC. This schematic depicts the four main TNBC subtypes under the TNBCtype-4 classification, highlighting their biological pathways and corresponding therapies. Basal-Like 1 (BL1, ~35%) exhibits increased cell-cycle and DNA-damage response, suggesting sensitivity to platinum chemotherapy and other DNA-damaging agents. Basal-Like 2 (BL2, ~22%) exhibits active growth factor signaling, making it a potential target for mTOR inhibitors. The Mesenchymal (M, ~27%) subtype drives epithelial-to-mesenchymal transition (EMT); therapies may target EMT or cancer stem cells (CSCs). Luminal Androgen Receptor (LAR, ~16%) is linked to androgen receptor (AR) signaling, suggesting potential for anti-androgen therapy. BL1: basal-like 1, BL2: basal-like 2, M: mesenchymal, LAR: luminal androgen receptor, EMT: epithelial-to-mesenchymal transition, CSCs: cancer stem cells.
TNBC’s heterogeneity spans clinical, histopathological, and molecular features, marked by high genomic instability and mutation rates (13, 23, 24). TNBC has a greater tumor mutational burden (25), which increases neoantigen production and the chances of immune detection (26). Yet, immunotherapy for TNBC is limited by low immunogenicity and an immunosuppressive tumor microenvironment (TME) (27). These features increase both neoantigen generation and immunogenicity, suggesting potential for immunotherapy (28, 29). However, complexity makes effective treatment strategies challenging. The TME heavily influences TNBC’s progression, immune evasion, and treatment resistance (14, 15). It is a complex, dynamic environment of diverse cellular and non-cellular components that interact closely with tumor cells (17, 30, 31). The TME contains immune cells like tumor-associated macrophages (TAMs), regulatory T cells (Tregs), tumor-infiltrating lymphocytes (TILs), and myeloid-derived suppressor cells (MDSCs), together with stromal cells, cancer-associated fibroblasts (CAFs), endothelial cells, extracellular matrix (ECM), and factors like cytokines, chemokines, and growth factors (32-39) (Figure 2). TNBC TME is especially heterogeneous, with an immunosuppressive profile that promotes tumor growth and treatment resistance (34, 40, 41).
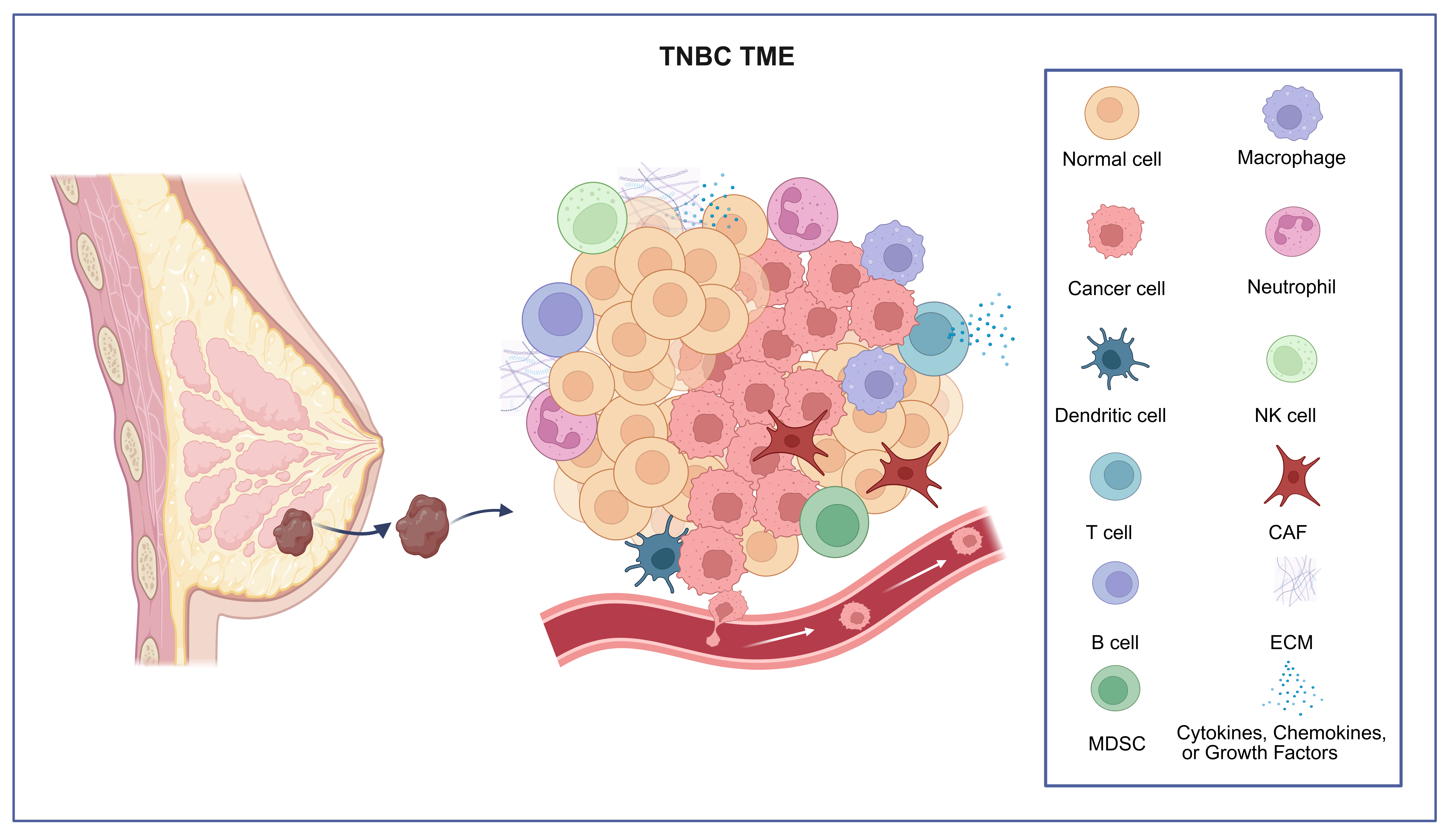
Figure 2: Components of the TNBC TME. The TNBC TME is a diverse ecosystem that tumor cells use to promote growth and evade immunity. It includes many cellular and non-cellular elements, creating an immunosuppressive environment. Components are macrophages, CAFs, ECM, MDSCs, T cells, B cells, DCs, NK cells, neutrophils, and soluble factors like cytokines, chemokines, and growth factors. TNBC: triple-negative breast cancer, TME: tumor microenvironment, CAF: cancer-associated fibroblast, ECM: extracellular matrix, MDSC: myeloid-derived suppressor cell, DC: dendritic cell, NK: natural killer. Figure created using BioRender.
Given the limited conventional treatments and the TME’s central role, research now focuses on targeting the TME to enhance anti-tumor immunity and overcome resistance (3, 15). Immunotherapies, especially immune checkpoint inhibitors (ICIs), are promising for restoring anti-tumor immune responses and are changing care in early and metastatic TNBC (15, 28, 42). However, not all patients respond to ICIs (response rate 5–23%) (43-47). Resistance remains a significant challenge; therefore, a better understanding of the TME and the development of combination approaches are crucial for improving therapy. New strategies target TME components such as TAMs, CAFs, and MDSCs, or target the ECM, inhibit angiogenesis, or alter tumor metabolism, often in combination with immunotherapy or chemotherapy (38, 48-51).
This review utilizes the TNBCtype-4 molecular subtyping system, which categorizes TNBC into four tumor-intrinsic subtypes: basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal (M), and luminal androgen receptor (LAR). This classification represents a refinement of the earlier TNBC-type system. The rationale for this consolidation is supported by histopathological analyses, which revealed that the transcriptional profiles of the previously defined immunomodulatory (IM) and mesenchymal stem-like (MSL) subtypes were not derived from the tumor epithelium (9). Instead, the IM signature was predominantly attributable to infiltrating lymphoid cells, while the MSL signature originated from tumor-associated stromal cells. Consequently, the TNBC-type-4 system provides a more accurate representation of tumor cell-specific biology, making it a robust framework for analyzing how intrinsic cancer cell pathways dictate interactions with the surrounding tumor microenvironment and influence therapeutic vulnerabilities.
2. Understanding the Tumor Microenvironment in Triple-Negative Breast Cancer
The TME in TNBC is highly complex and heterogeneous, exerting a significant influence on tumor behavior and therapeutic response (13, 14, 52, 53). Compared to hormone receptor-positive or HER2-positive breast cancers, TNBC demonstrates more extensive interactions with its microenvironment, marked by abundant immune cell infiltration and a dynamic stromal compartment (15, 28, 54). This heterogeneity extends beyond genetic and molecular differences within tumor cells to include the diverse composition and spatial organization of TME components (13, 52, 55, 56). A comprehensive understanding of this complexity is essential for the development of effective targeted therapies.
2.1 Cellular Components of the TNBC TME
The cellular landscape of the TNBC TME encompasses a range of immune and stromal cell populations that interact extensively with cancer cells. These interactions can either promote or inhibit tumor growth, significantly affecting therapeutic outcomes.
2.1.1 Tumor-Associated Macrophages (TAMs)
TAMs are among the most abundant immune cell populations in the TNBC TME and play critical, often dual, roles in tumor progression (17, 37, 41, 57, 58). Macrophages are highly plastic and can polarize into different functional phenotypes, broadly classified as M1 (pro-inflammatory, anti-tumor) and M2 (anti-inflammatory, pro-tumor) (17, 37, 41, 59, 60). In the TNBC TME, TAMs are frequently skewed towards the M2 phenotype, promoting tumor growth, angiogenesis, metastasis, and immunosuppression (57, 61). This polarization triggers signaling pathways that further reinforce the same polarized state. For example, cytokines such as IL-4 and IL-13 activate the JAK1/STAT6 pathway (62, 63), while IL-10 activates JAK1/STAT3 signaling (64), leading to the transcriptional upregulation of typical M2 markers, including Arginase 1, CD206, and immunosuppressive factors (65). The PI3K/Akt pathway is also a central node that integrates various signals to promote M2 survival and function (66). Studies highlight the intricate crosstalk between TNBC cells and TAMs. For instance, M2-type TAMs promote cancer stemness in TNBC cells by secreting vascular endothelial growth factor A (VEGFA) (17). Conversely, TNBC cells educated by TAMs exhibit elevated VEGFA, which further regulates macrophage polarization, forming a positive feedback loop that strengthens the cancer stem cell (CSC) phenotype via the VEGFA/NRP-1/GAPVD1/Wnt/β-catenin pathway (17).
This study underscores the pro-tumorigenic role of TAMs and the importance of this specific signaling axis in promoting stemness and potentially contributing to an immunosuppressive TME (Figure 3). The persistent activation of these intracellular networks, such as STAT3 and β-catenin, not only maintains the M2 phenotype but also establishes a feed-forward loop that reinforces the immunosuppressive TME (65, 67).The microenvironment of tumor occurrence is particularly rich in CD163+ macrophages, which is associated with poor survival prognosis (68-70). An eNAMPT/Ac-STAT3/DIRAS2 axis was identified in TAM-TNBC cell crosstalk, and CD163+ M2-like TAMs are relevant to unfavorable prognosis in TNBC (57). TNBC cell-conditioned medium induces M2 polarization, and these TAMs secrete eNAMPT, which activates STAT3 in TNBC cells via CCR5, downregulates the cancer suppressor DIRAS2, and increases CCL2 secretion. This creates a feedback loop in which CCL2 recruits more macrophages, thereby perpetuating the pro-tumorigenic environment (57). The eNAMPT-mediated activation of STAT3 is a prime example of a core intracellular signaling network driving tumor progression (57). The polarization of TAMs and the related signaling axes is shown in Figure 3.
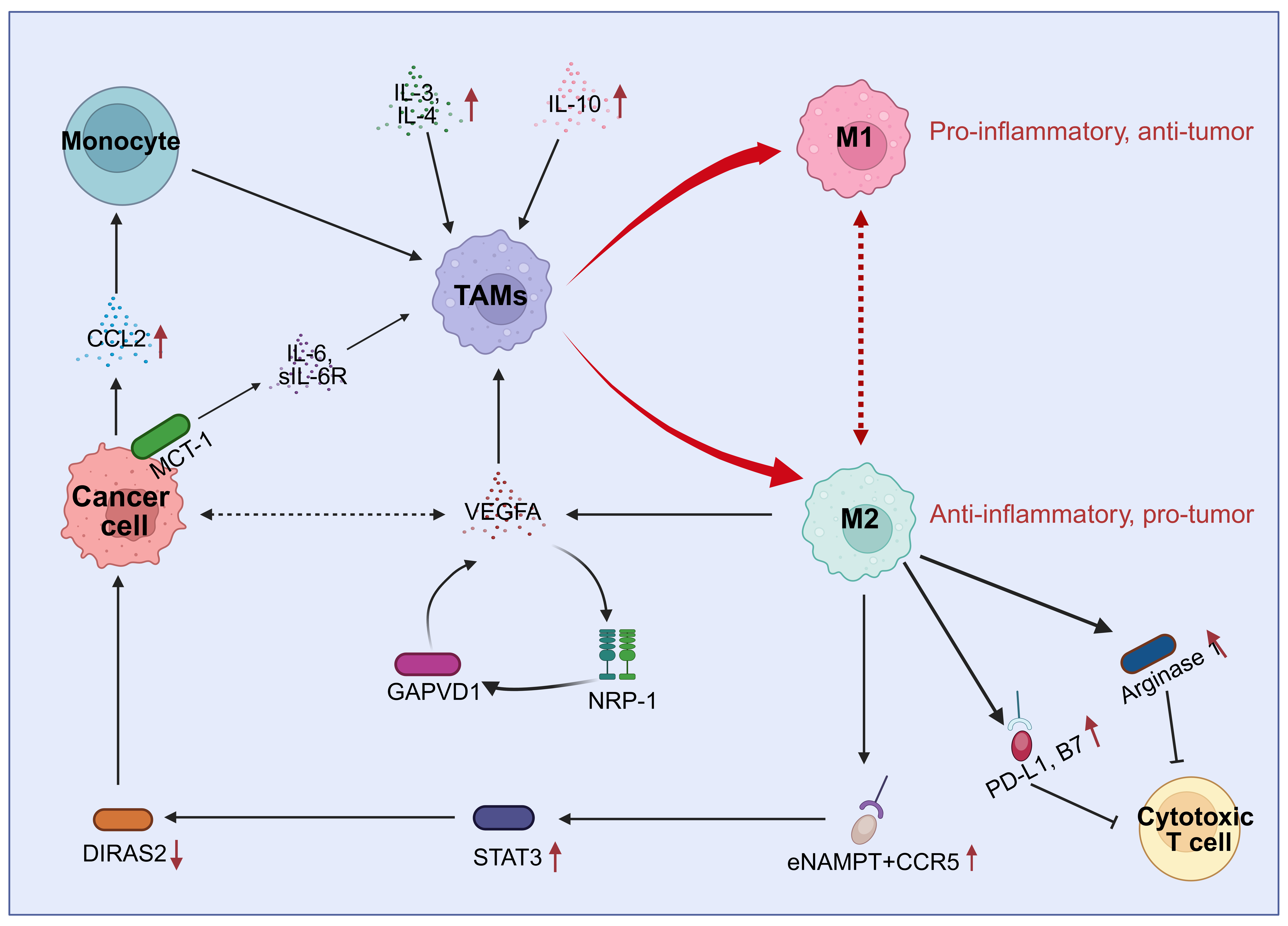
Figure 3: Signaling networks driving M2 polarization of TAMs and their pro-tumorigenic crosstalk with TNBC cells. Key pathways inducing M2 polarization. Cytokines in the TNBC TME, such as IL-4, IL-13, and IL-10, drive macrophage polarization towards an immunosuppressive M2 phenotype through activation of the JAK1/STAT6 and JAK1/STAT3 signaling axes, respectively. Core feedback loops between M2 TAMs and TNBC cells. Bidirectional crosstalk establishes self-reinforcing, pro-tumorigenic circuits: (i) The VEGFA-Stemness Loop: M2 TAM-derived VEGFA enhances cancer stemness in TNBC cells via the VEGFA/NRP-1/GAPVD1/Wnt/β-catenin pathway. TNBC cells reciprocally upregulate VEGFA, further polarizing TAMs. (ii) The eNAMPT/CCL2-Recruitment Loop: TNBC-conditioned TAMs secrete eNAMPT, which binds CCR5 on TNBC cells to activate STAT3, downregulate the tumor suppressor DIRAS2, and upregulate CCL2 secretion. CCL2 recruits fresh monocytes from the circulation, perpetuating the pool of TAMs for polarization. (iii) The IL-6-Polarization Loop: The oncogene MCT-1 in TNBC cells promotes the secretion of IL-6 and soluble IL-6R (sIL-6R), which accelerates M2 polarization. M2 TAMs, in turn, enhance the invasive potential and stemness of TNBC cells. Immunosuppressive mechanisms. M2 TAMs suppress cytotoxic T cell function through multiple mechanisms, including the expression of immune checkpoint ligands (PD-L1, B7) and metabolic depletion of L-arginine via Arginase 1 activity. TAMs: tumor-associated macrophages, TNBC: triple-negative breast cancer. This figure was created using BioRender.
The interaction between TNBC cells and TAMs also involves the release of inflammatory cytokines. The oncogene MCT-1 accelerates the polarization of M2-like macrophages by promoting interleukin-6 (IL-6) secretion from TNBC cells (37). This M2 polarization, in turn, enhances the invasive potential of TNBC cells. Furthermore, MCT-1 upregulates soluble IL-6 receptor (sIL-6R) levels, and targeting the IL-6/IL-6R axis effectively suppresses M2 polarization and TNBC stemness (37). The immunosuppressive nature of TAMs represents a significant barrier to effective immunotherapy. Mechanistically, TAMs suppress T cell activity by engaging immune checkpoints, such as PD-L1/PD-1 and CTLA-4/B7, and by metabolically depleting essential amino acids, including L-arginine, through the expression of Arginase 1, a downstream target of STAT3 signaling (71-73). Reprogramming TAMs from an immunosuppressive phenotype to an immune-activating state has been demonstrated to markedly enhance the therapeutic effect of immune checkpoint inhibitors in metastatic TNBC (38).
2.1.2 Cancer-Associated Fibroblasts (CAFs)
CAFs are another significant stromal cell population in the TNBC TME, contributing significantly to ECM remodeling, angiogenesis, immune suppression, and therapeutic resistance (49, 51, 74). CAFs secrete various growth factors, cytokines, and ECM components that create a supportive niche for tumor cells and influence the behavior of other TME components (49, 51, 75, 76). Distinct signaling pathways control the activation and tumor-promoting roles of CAFs. Among these, the TGF-β/SMAD axis is a key regulator, inducing α-SMA expression, a hallmark of CAFs, and stimulating extensive ECM synthesis and remodeling, which contribute to fibrosis and increased tissue rigidity (77-80). This stiffness, in turn, can further activate pro-survival signaling pathways, such as PI3K/Akt, in cancer cells (81).
Epithelial Membrane Protein 1 (EMP1) expression in TNBC cells positively correlates with stromal scores and CAF infiltration. Depletion of EMP1 in TNBC cells significantly inhibited CAF infiltration in vitro and in vivo (51). From a mechanistic perspective, the knockdown of IL-6 secretion from TNBC cells via the NF-κB pathway hinders CAF proliferation and inhibits TNBC progression and metastasis (51). CAFs also contribute to the physical barrier within the TME and secrete factors, such as transforming growth factor-beta (TGF-β), which promote fibrosis and immunosuppression (49). Beyond TGF-β, CAF-derived exosomes and soluble factors can activate oncogenic pathways in TNBC cells, including the Sonic Hedgehog (SHH) and Hippo/YAP/TAZ pathways, which are critically involved in promoting cancer cell stemness, proliferation, and resistance to chemotherapy (82, 83). Furthermore, CAFs contribute to immunosuppression by secreting cytokines, such as CXCL12, which can exclude T cells from the tumor core, and by expressing enzymes such as indoleamine 2,3-dioxygenase (IDO), which depletes tryptophan and suppresses T cell function (84-86).
2.1.3 Tumor-Infiltrating Lymphocytes (TILs)
TILs, especially cytotoxic T lymphocytes (CTLs), play a vital role in the anti-tumor immune response. The presence and density of TILs in TNBC are frequently associated with a superior prognosis and response to chemotherapy and immunotherapy (15, 28). TNBC is typically characterized by a higher density of TILs in comparison to other breast cancer subtypes, making it potentially more amenable to immunotherapy (15, 28, 87). However, the TME can render these TILs dysfunctional or exclude them from the tumor core, leading to an “immune cold” phenotype despite the presence of lymphocytes (88-90). The immunomodulatory subtype of TNBC is correlated with the highest expression of adaptive immune-related gene signatures. It exhibits a “fully inflamed” spatial pattern, positioning it as the most likely candidate for ICI response (52). In contrast, other subtypes, such as mesenchymal stem-like and luminal androgen receptor subtypes, often exhibit an immunosuppressive or “immune cold” phenotype characterized by stromal and metabolic features, as well as a “margin-restricted” spatial pattern of immune infiltration (52). Mechanisms of immune suppression in the TNBC TME can impair TIL function. C-terminal subunit of MUC1 (MUC1-C), a protein overexpressed in TNBC, links the activation of the interferon-gamma (IFN-γ) pathway to the suppression of the tumor immune microenvironment. MUC1-C activates the JAK1-STAT1-IRF1 signaling pathway, which induces immunosuppressive effectors such as IDO1 and COX2, and is related to the exhaustion and dysfunction of CD8+ T cells (88). This suggests that MUC1-C contributes to the ‘cold’ phenotype and is a promising target for enhancing ICI efficacy (88).
2.1.4 Myeloid-Derived Suppressor Cells (MDSCs)
MDSCs are a heterogeneous population of immature myeloid cells that expand abnormally during cancer and other pathological conditions characterized by chronic inflammation. Within the TME and the peripheral circulation of cancer patients, MDSCs accumulate in large numbers and exert potent immunosuppressive effects (91, 92). These cells can be broadly classified into two main subsets: polymorphonuclear (PMN-MDSCs), which share phenotypic similarities with neutrophils, and monocytic (M-MDSCs), which resemble monocytes but possess distinct transcriptional and functional profiles. Functionally, MDSCs employ multiple mechanisms to inhibit antitumor immunity. They suppress T cell proliferation and cytotoxic activity by producing reactive oxygen species (ROS), nitric oxide (NO), and arginase-1, which deplete essential amino acids, such as L-arginine and L-cysteine, from the local environment. MDSCs also promote the expansion and activation of Tregs and impair antigen presentation by dendritic cells, further weakening the adaptive immune response. In addition to their immunosuppressive roles, MDSCs contribute to tumor progression by secreting proangiogenic factors such as VEGF and MMP9, enhancing neovascularization and facilitating tumor cell invasion and metastasis. Through these combined mechanisms, MDSCs play a central role in shaping an immunosuppressive TME that enables tumor immune evasion and fosters resistance to various forms of immunotherapy, including immune checkpoint blockade and adoptive T cell transfer (91). Targeting MDSC recruitment, differentiation, or suppressive function is therefore an emerging therapeutic strategy to restore antitumor immunity and improve responses to immunotherapy.
2.1.5 Regulatory T cells (Tregs)
Tregs represent a specialized subset of CD4⁺ T lymphocytes that play a pivotal role in maintaining immune homeostasis and self-tolerance by suppressing excessive or autoreactive immune responses. The discovery of Tregs and their master transcription factor, FoxP3, a finding recognized by the 2025 Nobel Prize in Physiology or Medicine, highlighted their indispensable role in preventing autoimmunity and revealed their capacity to constrain antitumor immunity when co-opted by the TME. In the context of cancer, Tregs are frequently enriched within the TME, where they suppress cytotoxic T lymphocyte (CTL) activity and dampen the function of natural killer (NK) cells, dendritic cells, and other effector immune populations (34, 93-95). These suppressive effects are mediated by multiple mechanisms, including the secretion of inhibitory cytokines (e.g., IL-10, TGF-β, IL-35), the expression of immune checkpoint molecules (e.g., CTLA-4 and PD-1), and the consumption of essential growth factors such as IL-2, which deprives effector T cells of proliferative signals. In addition, Tregs can induce metabolic suppression via adenosine production and modulate antigen-presenting cell (APC) function, further reinforcing local immunosuppression. Accumulating evidence indicates that elevated Treg infiltration within tumors correlates with poor clinical outcomes and reduced responsiveness to ICIs, particularly in aggressive subtypes such as TNBC (34, 93).
Advanced transcriptomic analyses, including single-cell RNA sequencing and weighted gene co-expression network analysis (WGCNA), have identified gene modules associated with Treg infiltration, demonstrating that high Treg-related scores are predictive of unfavorable prognosis and diminished response to anti-PD-1 immunotherapy (93). Moreover, recent studies have uncovered intriguing interactions between the tumor microbiome and Treg-mediated immune modulation. For instance, Sphingobacterium multivorum colonization in TNBC tumors has been shown to accelerate tumor progression and impair anti-PD-1 efficacy, a process linked to increased Treg accumulation and concurrent depletion of CD8⁺ T cell infiltration (34). These findings underscore the multifaceted role of Tregs as key mediators of immune evasion and resistance to immunotherapy. Consequently, therapeutic strategies aimed at modulating Treg recruitment, stability, or suppressive activity, such as selective depletion within the TME or blockade of Treg-associated checkpoints like cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and TIGIT, are actively explored to enhance antitumor immune responses and improve the efficacy of current immunotherapeutic regimens.
2.1.6 Other Immune Cells
Beyond the major populations, other immune cells also contribute to the complexity of the TNBC TME. These include dendritic cells (DCs), B cells, neutrophils, and NK cells (13, 35, 96-99). Their roles are multifaceted and can be either pro- or anti-tumorigenic, depending on their phenotype and the specific context of the TME. DCs play a vital role in stimulating adaptive immune responses by presenting tumor antigens to T cells (35, 100). However, the TME can promote the development of tolerogenic DCs (tol-DCs) that suppress anti-tumor immunity (35). Tumors and associated immune cells in TNBC exhibit elevated CD74 expression (35). CD74 expressed on CD11c cells is critical in regulating tumor progression by mediating cross-talk between tumor-infiltrating tol-DCs and regulatory B cells (Bregs) (35). Neutrophils, another myeloid cell type, can also contribute to the immunosuppressive environment, and strategies to modulate their phenotype are being investigated (91). B cells, including Bregs, play a pivotal role in modulating the immune response within the TME (35).
2.2 Non-Cellular Components of the TNBC TME
The non-cellular components of the TNBC TME, including the ECM, soluble factors, and metabolic cues, form the physical and biochemical environment that supports cancer cells and influences cellular interactions.
2.2.1 Extracellular Matrix (ECM)
ECM is a complex network of proteins, glycoproteins, and proteoglycans that not only provides structural support to tissues but also actively regulates cellular behavior through biochemical and mechanical signaling (49, 101). By interacting with cell surface receptors such as integrins and syndecans, the ECM modulates processes including cell proliferation, migration, differentiation, and survival. In addition, the ECM can act as a physical and biochemical barrier, limiting immune-cell infiltration and impeding the penetration of therapeutic agents, including chemotherapeutics and monoclonal antibodies. In TNBC, the ECM is frequently subject to extensive remodeling, resulting in a stiff, dense, and highly cross-linked matrix. This desmoplastic transformation is primarily driven by CAFs, which secrete elevated levels of collagen, fibronectin, laminin, and other ECM components, as well as enzymes such as lysyl oxidase (LOX) and matrix metalloproteinases (MMPs) that remodel and stiffen the matrix (49, 102, 103). The altered ECM architecture not only enhances tumor cell invasion and metastatic potential but also generates elevated interstitial pressure, which can collapse blood vessels and reduce drug delivery. Furthermore, the stiffened ECM promotes mechanotransduction signaling in cancer cells, activating pathways such as YAP/TAZ, FAK, and PI3K/AKT, which contribute to tumor growth, survival, and therapy resistance.
Overall, ECM remodeling in TNBC establishes a tumor-promoting microenvironment that supports malignant progression, facilitates immune evasion, and reduces the efficacy of conventional and targeted therapies, highlighting the ECM as both a prognostic indicator and a therapeutic target (49).
2.2.2 Cytokines, Chemokines, and Growth Factors
Soluble factors within the TME, including vascular endothelial growth factor (VEGF), TGF-β, and IL-6, are secreted by tumor cells, immune cells, and stromal cells, establishing complex communication networks that regulate tumor progression, angiogenesis, and immune evasion (17, 57, 104, 105). These molecules mediate dynamic crosstalk among diverse cell populations, including tumor-associated macrophages, cancer-associated fibroblasts, T cells, and endothelial cells. This interaction shapes tumor architecture, promotes the survival and proliferation of malignant cells, and modulates the immune response, either suppressing or enhancing anti-tumor activity. In addition to cytokines, chemokines such as CXCL8 (IL-8) and CCL2 (MCP-1) play critical roles in recruiting immunosuppressive cells, promoting angiogenesis, and facilitating metastatic dissemination in TNBC (32, 57, 88, 106). Other soluble mediators, including tumor necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-γ), further influence the TME by regulating immune cell activation, polarization, and trafficking, as well as modulating the expression of adhesion molecules and matrix-remodeling enzymes. Collectively, these factors create a finely tuned, yet highly adaptable, network that governs tumor biology, orchestrates the interplay between immune suppression and activation, and contributes to therapeutic resistance in TNBC.
2.2.3 Metabolic Reprogramming and Hypoxia
Metabolic reprogramming is a defining feature of cancer cells, enabling them to meet the heightened energy and biosynthetic demands of rapid proliferation and survival under stress conditions. Within the TME, metabolic crosstalk between cancer cells and surrounding stromal and immune cells plays a critical role in shaping tumor evolution and modulating immune responses (107). In TNBC, tumor cells frequently exhibit enhanced glycolytic activity, known as the Warburg effect, even in the presence of sufficient oxygen (107). This high glycolytic flux not only generates ATP and biosynthetic precursors required for rapid growth but also leads to lactate accumulation, thereby acidifying the TME. The acidic and nutrient-depleted TME profoundly affects immune cell function. Effector T cells, NK cells, and dendritic cells often experience impaired proliferation, cytokine production, and cytotoxic activity in this environment. In contrast, immunosuppressive populations, such as Tregs and MDSCs, can proliferate (40). Metabolic competition for glucose, amino acids, and other metabolites between cancer and immune cells exacerbates immune dysfunction. Beyond glycolysis, TNBC cells also exhibit alterations in lipid metabolism, glutaminolysis, and oxidative phosphorylation, creating additional layers of metabolic adaptation that support tumor survival, metastasis, and resistance to therapy.
Thus, the interplay between cancer cell metabolism and the TME establishes a metabolic barrier to effective antitumor immunity, highlighting potential therapeutic opportunities to target metabolic pathways in TNBC.
2.2.4 Intratumoral Bacteria
The intratumoral microbiota is now recognized as a key modulator of the TME, with microbial dysbiosis significantly influencing cancer progression and therapeutic response (34, 108-110). Beyond its role in tumorigenesis, the microbiome plays a critical role in determining therapeutic efficacy. It can drive chemoresistance through mechanisms such as Fusobacterium nucleatum-induced protective autophagy and Gammaproteobacteria-mediated inactivation of gemcitabine (111). Similarly, immunotherapy outcomes are profoundly shaped by the microbiota, which can either enhance antitumor T-cell activity or promote immunosuppression through cytokine signaling pathways. The impact of specific bacteria is often context-dependent. For example, F. Nucleatum contributes to tumor development in various cancers through chronic inflammation, immune evasion, and direct cellular interactions. In colorectal and esophageal cancers, it induces autophagy-linked chemoresistance, while in breast cancer, it accelerates progression by reducing T-cell infiltration into the TME (112, 113). Another key player, enterotoxigenic Bacteroides fragilis, can colonize breast tissue and promote hyperplasia, growth, and metastasis (114). Notably, anticancer treatments can reciprocally reshape the tumor microbiome. Chemotherapy administration significantly alters the breast tumor microbiome, enriching for specific genera, such as Pseudomonas. The increased abundance of Brevundimonas and Staphylococcus in primary tumors is associated with the development of distant metastases, suggesting a potential link between therapy-induced microbial shifts and tumor recurrence (115).
2.2.5 Other Non-Cellular Factors
Various other molecules and pathways contribute to the complexity of the TNBC TME. RNA methylation modifications play a critical role in mediating cellular subtypes and influencing prognosis and immune therapy response in TNBC (116). Targeting Long non-coding RNAs, such as MALAT1, can alter the immune microenvironment, thereby reducing immunosuppression and increasing T-cell infiltration (117). Thymidine Kinase-1 expression is associated with high Treg-cell infiltration and poor prognosis, and may serve as a biomarker and target (93, 118). Lymphocyte activation gene-3, an immune checkpoint protein, is highly expressed in TILs in TNBC and correlates with the ligand programmed death-ligand 1 (PD-L1), suggesting its potential as a target for immunotherapy. However, its prognostic significance requires further investigation (119). CXCL16 and STAT1 signaling in myeloid cells are implicated in immune suppression and resistance to chemotherapy-primed ICI therapy, with STAT1 inhibition showing potential to sensitize TNBC to immune checkpoint blockade (36).
The intricate interplay between cellular and non-cellular components generates a distinct and challenging microenvironment in TNBC. TME heterogeneity complicates the development of effective therapeutic strategies. Tumors may be classified as “hot” (inflamed, high TILs) or “cold” (non-inflamed, low TILs), yet even “hot” tumors can exhibit dysfunctional or excluded immune cells (88, 89). Addressing this complexity requires targeted interventions that modulate specific TME components or pathways to shift the balance toward anti-tumor immunity and overcome resistance.
3. Targeting Approaches to Overcome Immune Escape in the Triple-Negative Breast Cancer Tumor Microenvironment
The efficacy of cancer immunotherapy, particularly ICIs, is profoundly limited by the capacity of tumors to enact a multitude of immune escape mechanisms. These mechanisms, driven by cellular components such as TAMs, MDSCs, and Tregs, as well as physical barriers such as fibrotic ECM and TME metabolic pathways, create an immunosuppressive niche that excludes or inactivates cytotoxic immune cells. Therefore, the next frontier in TNBC therapy lies in developing strategies that build upon the foundation of ICIs by not only unleashing anti-tumor immunity but also systematically dismantling these immune escape pathways. This section reviews therapeutic approaches framed through the lens of overcoming specific immune evasion strategies (Figure 4).
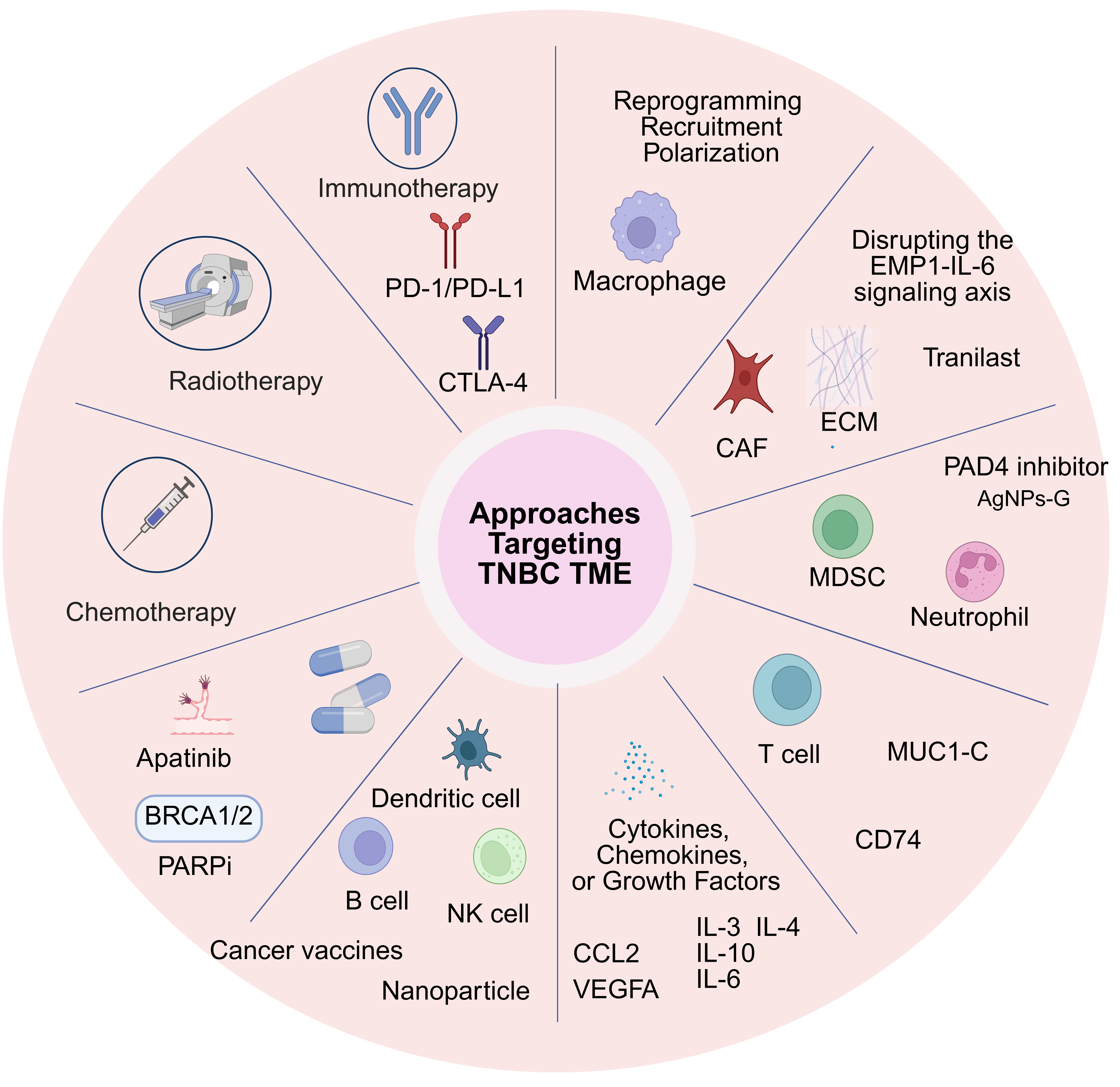
Figure 4: Overview of therapeutic strategies targeting the TNBC TME. Emerging treatments aim to overcome immunosuppression by targeting key components of the TME. Strategies illustrated include: 1. Immune checkpoint inhibitors (e.g., anti-PD-1 and anti-CTLA-4) to activate T cells; 2. Agents targeting TAMs and MDSCs, and other cells for depletion or reprogramming; 3. Approaches to modulate cancer-associated fibroblasts (CAFs) and the extracellular matrix (ECM); 4. Inhibition of critical cytokine and chemokine signaling networks (e.g., IL-3, IL-4, IL-10, IL-6, and CCL2); 5. Conventional cytotoxic therapies: chemotherapy and radiotherapy; 6. Other novel cell-based therapies. TNBC: triple-negative breast cancer, TME: tumor microenvironment, TAMs: tumor-associated macrophages, MDSCs: myeloid-derived suppressor cells, CAFs: cancer-associated fibroblasts, ECM: the extracellular matrix. This figure was created using BioRender.
3.1 Immune Checkpoint Inhibitors (ICIs)
ICIs, especially those targeting programmed cell death protein 1 (PD-1) and PD-L1, have shown significant promise in TNBC (14, 48, 120-122). Compared to other breast cancer subtypes, TNBC exhibits superior immunogenicity, characterized by a higher mutation burden and TIL infiltration, making it a suitable candidate for immunotherapy (13, 15, 28). ICIs function by blocking inhibitory signals that hinder T cells from targeting cancer cells and restoring or enhancing anti-tumor immunity (13, 15, 28). The limitations of single-agent ICIs underscore the need for combination strategies. Simultaneously targeting immune checkpoints and modulating the TME can enhance anti-tumor immunity and overcome resistance mechanisms. Clinical evidence supports the use of PD-1/PD-L1 inhibitors in TNBC across early and metastatic stages (123, 124). Still, their efficacy is limited to a subset of patients due to the complex and often immunosuppressive TNBC TME (28, 40, 48). Factors influencing ICI response include the level and spatial distribution of TILs, PD-L1 expression on tumor and immune cells, and the presence of immunosuppressive cell populations and pathways within the TME (28, 52, 88). For example, the immunomodulatory subtype of TNBC, characterized by a “fully inflamed” TME, appears to be the most responsive to ICIs (52). Conversely, the recruitment and activation of immunosuppressive cell populations are primary mediators of ICI resistance. For instance, Tregs directly suppress CTL activity in the TME, while M2-polarized TAMs and MDSCs establish a localized immunosuppressive milieu through cytokine secretion and metabolic dysregulation, such as arginase and IDO (65, 85). Furthermore, the CAF-derived dense ECM creates a physical barrier that impedes T cell infiltration (49, 101). This underscores the need for combinatorial strategies that target these specific escape mechanisms to overcome ICI resistance. In addition to PD-1/PD-L1, other immune checkpoints are being investigated as therapeutic targets in TNBC. Lymphocyte activation gene-3 is another inhibitory receptor expressed on TILs. Its high expression in the TNBC TME, often associated with PD-L1, suggests that dual blockade may benefit certain patients (119). CTLA-4 is a well-established immune checkpoint target, and the combination of anti-CTLA-4 with anti-PD-1 has shown performance in other cancers (49, 107).
3.2 Strategies to Counteract Immune Escape Mechanisms
3.2.1 Reprogramming Myeloid-Driven Immunosuppression
To counteract myeloid-driven immunosuppression, a primary immune escape pathway, several strategies targeting TAMs and MDSCs are in development. Given their prominent role in promoting tumor growth, metastasis, and immunosuppression, TAMs are attractive therapeutic targets in TNBC. Strategies include inhibiting TAM recruitment, depleting TAMs, or reprogramming their phenotype from pro-tumorigenic M2 to anti-tumorigenic M1 (17, 37, 41, 50, 125-127). Targeting the VEGFA/NRP-1/GAPVD1 axis, which facilitates crosstalk between TNBC cells and TAMs and enhances cancer stemness, represents a potential therapeutic strategy (17). Targeting the eNAMPT/CCR5/CCL2 feedback loop between TAMs and TNBC cells, which facilitates M2 polarization and macrophage recruitment, could disrupt the pro-tumorigenic niche (57). Blocking the IL-6/IL-6R axis, which drives M2 polarization and cancer stemness, is another promising approach (37). Using MEK, PPARγ, or HDAC inhibitors to block the MEK/PPARγ/RA signaling axis, which drives M2-type macrophage polarization, represents an encouraging treatment option for modulating the tumor microenvironment and enhancing anti-tumor immunity (128). Novel approaches employing targeted delivery systems are being developed that specifically reprogram TAMs to the M1 phenotype (41, 129, 130). Plant-derived extracellular vesicles (EVs) have also been shown to induce M1 polarization of TAMs, thereby contributing to their antitumor effects (50). A newly developed biomimetic tumor cell membrane-encapsulated nanodelivery system, assembled from a second near-infrared photothermal agent, chemotherapeutic drugs, and PD-L1 inhibitors coated with TNBC cell membranes, is used to enhance immunotherapy (120). Targeting signaling pathways within TAMs is also being explored. The PI3K-γ inhibitor Eganelisib was shown to reprogram TAMs from an immunosuppressive to an immune-activating phenotype, improving ICI effectiveness in metastatic TNBC (38). TAMs promote epithelial-to-mesenchymal transition (EMT) and strengthen the CSC characteristics in TNBC by activating the CCL2/AKT/β-catenin signaling pathway (131), which could offer novel approaches for diagnosing and treating TNBC. Targeting TAM-specific pathways to influence the TME provides a promising therapeutic strategy.
Targeting immunosuppressive cells such as MDSCs and Tregs to reduce their numbers or suppress their function is critical for enhancing anti-tumor immune responses (91, 93, 132). Targeting specific pathways within these cells, or the factors that recruit them, is a potential strategy. A novel peptidyl arginine deiminase 4 inhibitor has the potential to reshape the phenotype of neutrophils, a subset of MDSCs, and reduce MDSC accumulation (91).
3.2.2 Alleviating Treg-Mediated Inhibition
Strategies aimed at limiting Treg recruitment and suppressive activity represent a promising approach to enhancing antitumor immunity. Tumor cells can actively recruit Tregs through chemokine signaling; for example, CCL20, secreted by tumor cells under the influence of intra-tumoral bacteria, has been shown to promote Treg accumulation in the TME, facilitating immune evasion (34). Inhibiting such chemokine-mediated pathways could therefore reduce Treg-cell infiltration, restore effector T cell function, and increase tumor responsiveness to immunotherapy. In addition to chemokine-driven recruitment, specific molecular markers associated with Treg infiltration and poor clinical outcomes, such as Thymidine Kinase-1 (TK1), have emerged as potential therapeutic targets (93). Modulating these molecules may disrupt pro-tumorigenic interactions between Tregs and other TME components, thereby improving antitumor immune responses and patient outcomes. Moreover, the composition of the TME itself can be therapeutically remodeled to favor effector immune responses. Agents such as silver nanoparticle conjugates (AgNPs-G) have been shown to selectively reduce Treg populations while simultaneously promoting the infiltration and activation of cytotoxic T lymphocytes and other effector immune cells (32). Such strategies highlight the potential of combining TME modulation with direct targeting of Treg recruitment pathways to achieve stronger and more sustained antitumor immunity.
Collectively, these strategies underscore the importance of targeting both the molecular drivers of Treg recruitment and the broader immunosuppressive landscape of the TME as a multifaceted approach to overcome immune evasion in cancer.
3.2.3 Disrupting the Stromal Barrier
CAFs contribute to ECM remodeling, immune suppression, and drug resistance, making them relevant therapeutic targets (49, 51, 133). Strategies include inhibiting CAF recruitment or function and disrupting the CAF-mediated ECM barrier. Targeting EMP1 or disrupting the EMP1/IL-6 signaling axis could reduce CAF infiltration and impede tumor progression (51). Disrupting the ECM and reducing fibrosis, processes often driven by CAFs, can enhance drug delivery and improve immune cell infiltration (49). Inhibiting TGF-β, a key cytokine secreted by CAFs, using Tranilast has been shown to normalize the TME by reducing ECM components, improving perfusion, and facilitating immune cell infiltration (49). Targeting CAFs can also disrupt their role in forming cancer stem cell niches, potentially leading to improved treatment outcomes (134).
3.2.4 Reversing Metabolic Immune Suppression
Metabolic reprogramming in the TME contributes to immune suppression and resistance (135). Inhibiting glycolysis in TNBC cells, for example, by targeting GLUT1, can reduce immunosuppressive factors such as PD-L1 glycosylation and metabolically rewire Tregs, thereby enhancing the ICI response (107). Targeting GLUT3, which is elevated in metastatic TNBC and linked to glycolysis and the inflammatory TME, is another potential metabolic target (106). Modulating the NADPH pathway in tumor cells and the TME can influence redox balance and immune responses, as demonstrated by a nanomedicine that selectively depletes NADPH in tumor cells to enhance low-dose radiotherapy and anti-PD-L1 therapy (40). Inhibiting STAT1 signaling in myeloid cells, which is associated with an immunosuppressive state linked to chemotherapy priming, can sensitize TNBC to immune checkpoint blockade (36). It highlights the importance of targeting metabolic/signaling pathways in immune cells within the TME.
3.2.5 Novel and Emerging Targets
Numerous additional molecules and pathways within the TME are under investigation as therapeutic targets. Targeting the MUC1-C, which promotes immunosuppression by activating the IFN-γ pathway and depleting TILs, could improve ICI efficacy (88). Inhibiting CD74, which induces the expansion of tol-DCs and Bregs, offers a strategy to reverse immunosuppression mediated by these cell types (35). Modulating the AnxA1/FPR1 axis, which interacts with IL-6 signaling and affects TME components like fibroblasts, could also influence tumor progression (104). Targeting long non-coding RNAs, such as Malat1, can alter the immunosuppressive TME and increase T-cell infiltration (117). Inducing pyroptosis in TNBC cells using HDAC inhibitors can promote immune cell infiltration and enhance anti-cancer immunity (136). Targeting intratumoral bacteria, for instance, by killing F. nucleatum to release immunopotentiating pathogen-associated molecular patterns (PAMPs), represents a novel strategy to warm up ‘cold’ tumors and enhance immunotherapy (137).
3.3 Combination Approaches
Due to the complexity and redundancy of immunosuppressive mechanisms in the TNBC TME, single-agent therapies frequently demonstrate limited efficacy. Consequently, combination strategies that concurrently target multiple TME components or pathways, or integrate TME targeting with conventional therapies, are under active investigation to improve therapeutic efficacy and address treatment resistance (13, 28, 138, 139).
A cornerstone of this approach is the combination of ICIs with cytotoxic chemotherapy. Chemotherapy can modulate the TME to enhance ICI activity by inducing immunogenic cell death, releasing tumor antigens and damage-associated molecular patterns that stimulate anti-tumor immunity (137) (140). ICIs combined with chemotherapy have shown promising clinical benefit in metastatic and early TNBC (13, 48, 141, 142). However, chemotherapy can also induce immunosuppressive myeloid cells and alter the composition of the tumor microenvironment (36, 143), highlighting the need for rational combinations and timing.
Beyond chemotherapy, combining ICIs with anti-angiogenic agents represents another promising avenue. Anti-angiogenic therapy can reprogram the tumor microenvironment, rendering breast cancer more responsive to PD-1/PD-L1 blockade. Angiogenic factors promote immunosuppression by inhibiting the function of antigen-presenting and effector cells, which in turn drive angiogenesis, perpetuating a vicious cycle of immune dysfunction (144). A phase 2 trial combining Apatinib (a VEGFR2 inhibitor) with Sintilimab (anti-PD-1) and chemotherapy showed a high pathological complete response rate in early TNBC (48). The incorporation of Camrelizumab (anti-PD-1) and Apatinib demonstrates good tolerance in advanced TNBC, showing promising objective response rates and progression-free survival, regardless of treatment line or PD-L1 expression status (145).
The combination of ICIs with radiotherapy is also under active investigation. Radiation therapy can alter the TME by inducing immunogenic cell death and potentially reversing local immunosuppression, thereby acting as an in situ vaccine (14, 146-149). However, the mechanisms underlying radiation resistance and immune changes remain under investigation. Combining low-dose radiotherapy with anti-PD-L1 therapy using a nanomedicine that modulates immunometabolism shows promise in enhancing the efficacy of anti-PD-L1 treatment (40).
To enable these sophisticated combinations, nanomedicine and targeted delivery systems provide a versatile platform. Nanoparticles offer a versatile platform for co-delivering multiple therapeutic agents directly to the TME, overcoming biological barriers, enhancing specificity, and reducing systemic toxicity. These systems increase drug solubility, stability, and circulation time, thereby achieving targeted delivery via the enhanced permeability and retention effect or active targeting of TME components (32, 40, 49, 50, 120, 138, 139, 150-152). Furthermore, nanoparticles can be engineered to be responsive to the distinct properties of the TME, for instance, acidic pH, oxygen deprivation, or heightened enzyme activity, enabling controlled drug release (89, 107, 150).
The combination of ICIs with PARP inhibitors (PARPi) remains an active area of investigation. Preclinical and early clinical data continue to suggest that PARPi can enhance antitumor immunity by increasing PD-L1 expression, creating a rationale for this strategy. Preclinical studies reveal that PARPi exhibit dual immunomodulatory effects: while they can improve tumor immunogenicity by increasing neoantigen exposure and activating STING signaling, they may also upregulate compensatory immune checkpoints, such as PD-L1. However, combining PARPi with PD-1/PD-L1 blockade synergistically enhances T-cell-mediated tumor killing in vivo by overcoming this adaptive resistance (153-155). In a single-arm, open-label, phase 2 trial of niraparib combined with pembrolizumab for advanced or metastatic triple-negative breast cancer, the combination demonstrated a tolerable safety profile and promising antitumor activity, irrespective of BRCA mutation status (156). In BRCA-mutant patients, the MEDIOLA trial (Olaparib plus Durvalumab) achieved a disease control rate of 80% at 12 weeks and 50% at 28 weeks (157). These results confirm the clinical potential of this strategy. However, the phase III KEYLYNK-009 trial (2024), which evaluated niraparib plus pembrolizumab versus chemotherapy in unselected metastatic TNBC, did not meet its primary endpoints of progression-free survival and overall survival in the overall population. Nonetheless, improvements in both progression-free survival and overall survival were observed in patients with tumor BRCA mutations treated with pembrolizumab plus Olaparib, compared to those receiving pembrolizumab plus chemotherapy, suggesting a potential role for this combination as a maintenance strategy in this biomarker-defined subgroup (158).
Furthermore, other emerging multimodal approaches include combining ICIs with cancer vaccines or NK cell therapy (13, 159-163). Combining therapies that induce different forms of immunogenic cell death, such as immuno-chemodynamic therapy enhanced by targeting intratumoral bacteria, can synergistically activate anti-tumor immunity (137). Advanced preclinical models, such as 3D TME-on-a-chip and bioprinted tumor-stroma systems, are revolutionizing our approach to TNBC. They not only elucidate critical tumor-stromal-immune crosstalk but also serve as powerful platforms for evaluating novel therapies and combinations, particularly against therapy-resistant CSCs within their complex microenvironment (164, 165). Strategies that combine targeting CSCs with modulating the TME are also promising, as the TME provides a niche for CSCs and influences their behavior (166-169).
The diverse range of targeting approaches reflects the multifaceted nature of the TNBC TME. Strategies that aim to reprogram or disrupt key TME components, either alone or in combination with ICIs or conventional therapies, have significant potential to improve response rates and overcome treatment resistance. The development of targeted delivery systems, particularly TME-responsive nanoparticles, offers exciting opportunities to enhance the specificity and efficacy of these novel therapies.
Discussion
Despite advances in understanding the TNBC TME and in developing TME-targeted therapies, significant challenges persist. Different TNBC subtypes exhibit distinct TME profiles and varying responsiveness to therapies, emphasizing the need for personalized treatment approaches (52).
Mechanisms of resistance to TME-targeted therapies, including ICIs, are complex and involve multiple redundant pathways and cell populations (15, 28, 54, 170, 171). The plasticity of TME cells, or the dynamic interplay among different immunosuppressive components, can lead to treatment escape (17, 34, 38, 41, 51, 137). Furthermore, the physical barrier imposed by the dense ECM can limit drug penetration and immune cell infiltration, contributing to resistance (101). Metabolic adaptations and hypoxia within the TME also create unfavorable conditions for anti-tumor immunity and can promote resistance to various therapies, including radiation (14, 106, 172-174).
Identifying reliable predictive biomarkers is paramount for stratifying patients and selecting the most appropriate TME-targeted therapies or combinations. While PD-L1 expression is employed as a biomarker for ICI therapy, its predictive reliability is limited, and better markers are needed (15, 28, 175-177). Biomarkers reflecting the overall immune landscape, specific immunosuppressive pathways, or the presence of specific TME components are under investigation (15, 28, 34-36, 51, 88, 93, 119, 178, 179). RNA methylation regulators and oxeiptosis scores are also being explored for their prognostic and predictive potential (48, 180). The application of multi-omics approaches is highly significant for identifying biomarkers that reveal molecular characteristics and key pathways driving TNBC progression and influencing the TME (181, 182).
Future directions in targeting the TNBC TME involve several key areas. Further dissecting the intricate crosstalk between TME components and cancer cells is essential for identifying novel targets and understanding mechanisms of resistance. Developing strategies to reprogram immunosuppressive cells towards an anti-tumor phenotype effectively remains a priority. Targeting the ECM to improve drug delivery and immune infiltration is also crucial. Modulating the metabolic landscape of the TME to favor anti-tumor immunity is another promising avenue. The emerging role of the intratumoral microbiome suggests the development of novel therapeutic strategies. Combination therapies are likely to be the cornerstone of future treatment for TNBC. Rational design of combinations requires a thorough understanding of how different therapies affect the TME and interact with one another.
In conclusion, the TME is a pivotal determinant of TNBC progression, immune evasion, and therapeutic resistance. Targeting the TME, particularly through strategies that enhance anti-tumor immunity and overcome immunosuppression, offers promising avenues for improving TNBC treatment outcomes. While immune checkpoint inhibitors have demonstrated significant potential, their efficacy is often limited by the complex TME. Future efforts should focus on developing rational combination therapies that simultaneously target multiple pro-tumorigenic and immunosuppressive components of the TME. Guided by robust predictive biomarkers, the development of more effective and personalized treatments for TNBC patients can be achieved.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17-48. https://doi.org/10.3322/caac.21763
2. Arnold M, Morgan E, Rumgay H, Mafra A, Singh D, Laversanne M, et al. Current and future burden of breast cancer: global statistics for 2020 and 2040. Breast. 2022;66:15-23. https://doi.org/10.1016/j.breast.2022.08.010
3. Wu TN, Chen HM, Shyur LF. Current advancements of plant-derived agents for triple-negative breast cancer therapy through deregulating cancer cell functions and reprogramming tumor microenvironment. Int J Mol Sci. 2021;22(24):13571. https://doi.org/10.3390/ijms222413571
4. Pareja F, Reis-Filho JS. Triple-negative breast cancers - a panoply of cancer types. Nat Rev Clin Oncol. 2018;15(6):347-348. https://doi.org/10.1038/s41571-018-0001-7
5. Kwong A, Mang OWK, Wong CHN, Chau WW, Law SCK; Hong Kong Breast Cancer Research Group. Breast cancer in Hong Kong, Southern China: the first population-based analysis of epidemiological characteristics, stage-specific, cancer-specific, and disease-free survival in breast cancer patients: 1997-2001. Ann Surg Oncol. 2011;18(11):3072-3078. https://doi.org/10.1245/s10434-011-1960-4
6. Waks AG, Winer EP. Breast cancer treatment. JAMA. 2019;321(3):288-300. https://doi.org/10.1001/jama.2018.19323
7. Capuozzo M, Celotto V, Santorsola M, Fabozzi A, Landi L, Ferrara F, et al. Emerging treatment approaches for triple-negative breast cancer. Med Oncol. 2023;41(1):5. https://doi.org/10.1007/s12032-023-02257-6
8. Hwang KT, Kim J, Jung J, Chang JH, Chai YJ, Oh SW, et al. Impact of breast cancer subtypes on prognosis of women with operable invasive breast cancer: a population-based study using SEER database. Clin Cancer Res. 2019;25(6):1970-1979. https://doi.org/10.1158/1078-0432.Ccr-18-2782
9. Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One. 2016;11(6):e0157368. https://doi.org/10.1371/journal.pone.0157368
10. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750-2767. https://doi.org/10.1172/jci45014
11. Yin L, Duan JJ, Bian XW, Yu SC. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020;22(1):61. https://doi.org/10.1186/s13058-020-01296-5
12. Lips EH, Mulder L, Oonk A, Van der Kolk LE, Hogervorst FBL, Imholz ALT, et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer. 2013;108(10):2172-2177. https://doi.org/10.1038/bjc.2013.144
13. Kudelova E, Smolar M, Holubekova V, Hornakova A, Dvorska D, Lucansky V, et al. Genetic heterogeneity, tumor microenvironment and immunotherapy in triple-negative breast cancer. Int J Mol Sci. 2022;23(23):14937. https://doi.org/10.3390/ijms232314937
14. Niture S, Ghosh S, Jaboin J, Seneviratne D. Tumor microenvironment dynamics of triple-negative breast cancer under radiation therapy. Int J Mol Sci. 2025;26(6):2795. https://doi.org/10.3390/ijms26062795
15. Guo Z, Zhu Z, Lin X, Wang S, Wen Y, Wang L, et al. Tumor microenvironment and immunotherapy for triple-negative breast cancer. Biomark Res. 2024;12(1):166. https://doi.org/10.1186/s40364-024-00714-6
16. Wang X, Song Y, Yu L, Xue X, Pang M, Li Y, et al. Co-delivery of hesperetin and cisplatin via hyaluronic acid-modified liposome for targeted inhibition of aggression and metastasis of triple-negative breast cancer. ACS Appl Mater Interfaces. 2023;15(29):34360-34377. https://doi.org/10.1021/acsami.3c03233
17. Wang L, Zhang L, Zhao L, Shao S, Ning Q, Jing X, et al. VEGFA/NRP-1/GAPVD1 axis promotes progression and cancer stemness of triple-negative breast cancer by enhancing tumor cell-macrophage crosstalk. Int J Biol Sci. 2024;20(2):446-463. https://doi.org/10.7150/ijbs.86085
18. Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov. 2019;9(2):176-198. https://doi.org/10.1158/2159-8290.Cd-18-1177
19. Cortes J, Rugo HS, Cescon DW, Im SA, Yusof MM, Gallardo C, et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N Engl J Med. 2022;387(3):217-226. https://doi.org/10.1056/NEJMoa2202809
20. André F, Zielinski CC. Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents. Ann Oncol. 2012;23(Suppl 6):vi46–51. https://doi.org/10.1093/annonc/mds195
21. Lehmann BD, Abramson VG, Dees EC, Shah PD, Ballinger TJ, Isaacs C, et al. Atezolizumab in combination with carboplatin and survival outcomes in patients with metastatic triple-negative breast cancer: the TBCRC 043 phase 2 randomized clinical trial. JAMA Oncol. 2024;10(2):193-201. https://doi.org/10.1001/jamaoncol.2023.5424
22. Tarantino P, Corti C, Schmid P, Cortes J, Mittendorf EA, Rugo H, et al. Immunotherapy for early triple negative breast cancer: research agenda for the next decade. NPJ Breast Cancer. 2022;8(1):23. https://doi.org/10.1038/s41523-022-00386-1
23. Safonov A, Jiang T, Bianchini G, Gyorffy B, Karn T, Hatzis C, et al. Immune gene expression is associated with genomic aberrations in breast cancer. Cancer Res. 2017;77(12):3317-3324. https://doi.org/10.1158/0008-5472.Can-16-3478
24. Jain A, Barge A, Parris CN. Combination strategies with PARP inhibitors in BRCA-mutated triple-negative breast cancer: overcoming resistance mechanisms. Oncogene. 2025;44(4):193-207. https://doi.org/10.1038/s41388-024-03227-6
25. Derakhshan F, Reis-Filho JS. Pathogenesis of triple-negative breast cancer. Annu Rev Pathol. 2022;17:181-204. https://doi.org/10.1146/annurev-pathol-042420-093238
26. Liu Y, Hu Y, Xue J, Li J, Yi J, Bu J, et al. Advances in immunotherapy for triple-negative breast cancer. Mol Cancer. 2023;22(1):145. https://doi.org/10.1186/s12943-023-01850-7
27. Michaels E, Chen N, Nanda R. The role of immunotherapy in triple-negative breast cancer (TNBC). Clin Breast Cancer. 2024;24(4):263-270. https://doi.org/10.1016/j.clbc.2024.03.001
28. Loizides S, Constantinidou A. Triple negative breast cancer: immunogenicity, tumor microenvironment, and immunotherapy. Front Genet. 2023;13:1095839. https://doi.org/10.3389/fgene.2022.1095839
29. Huo W, Yang X, Wang B, Cao L, Fang Z, Li Z, et al. Biomineralized hydrogel DC vaccine for cancer immunotherapy: a boosting strategy via improving immunogenicity and reversing immune-inhibitory microenvironment. Biomaterials. 2022;288:121722. https://doi.org/10.1016/j.biomaterials.2022.121722
30. Tan Z, Kan C, Sun M, Yang F, Wong M, Wang S, et al. Mapping breast cancer microenvironment through single-cell omics. Front Immunol. 2022;13:868813. https://doi.org/10.3389/fimmu.2022.868813
31. Harris MA, Savas P, Virassamy B, O'Malley MMR, Kay J, Mueller SN, et al. Towards targeting the breast cancer immune microenvironment. Nat Rev Cancer. 2024;24(8):554-577. https://doi.org/10.1038/s41568-024-00714-6
32. Félix-Pina P, Franco Molina MA, García Coronado PL, Prado-Garcia H, Zarate-Trivino DG, Castro-Valenzuela BE, et al. β-D-glucose-reduced silver nanoparticles remodel the tumor microenvironment in a murine model of triple-negative breast cancer. Int J Mol Sci. 2024;25(15):8432. https://doi.org/10.3390/ijms25158432
33. Neophytou CM, Panagi M, Stylianopoulos T, Papageorgis P. The role of tumor microenvironment in cancer metastasis: molecular mechanisms and therapeutic opportunities. Cancers (Basel). 2021;13(9):2053. https://doi.org/10.3390/cancers13092053
34. Mai Z, Fu L, Su J, To KKW, Yang C, Xia C. Intra-tumoral sphingobacterium multivorum promotes triple-negative breast cancer progression by suppressing tumor immunosurveillance. Mol Cancer. 2025;24(1):6. https://doi.org/10.1186/s12943-024-02202-9
35. Pellegrino B, David K, Rabani S, Lampert B, Tran T, Doherty E, et al. CD74 promotes the formation of an immunosuppressive tumor microenvironment in triple-negative breast cancer in mice by inducing the expansion of tolerogenic dendritic cells and regulatory B cells. PLOS Biol. 2024;22(11):e3002905. https://doi.org/10.1371/journal.pbio.3002905
36. Palakurthi B, Fross SR, Guldner IH, Aleksandrovic E, Liu X, Martino AK, et al. Targeting CXCL16 and STAT1 augments immune checkpoint blockade therapy in triple-negative breast cancer. Nat Commun. 2023;14(1):2109. https://doi.org/10.1038/s41467-023-37727-y
37. Weng YS, Tseng HY, Chen YA, Shen PC, Al Haq AT, Chen LM, et al. MCT-1/mir-34a/il-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Cancer. 2019;18(1):42. https://doi.org/10.1186/s12943-019-0988-0
38. O’Connell BC, Hubbard C, Zizlsperger N, Fitzgerald D, Kutok JL, Varner J, et al. Eganelisib combined with immune checkpoint inhibitor therapy and chemotherapy in frontline metastatic triple-negative breast cancer triggers macrophage reprogramming, immune activation and extracellular matrix reorganization in the tumor microenvironment. J Immunother Cancer. 2024;12(8):e009160. https://doi.org/10.1136/jitc-2024-009160
39. Mayer S, Milo T, Isaacson A, Halperin C, Miyara S, Stein Y, et al. The tumor microenvironment shows a hierarchy of cell-cell interactions dominated by fibroblasts. Nat Commun. 2023;14(1):5810. https://doi.org/10.1038/s41467-023-41518-w
40. Wang Y, Gao D, Jin L, Ren X, Ouyang Y, Zhou Y, et al. NADPH selective depletion nanomedicine‐mediated radio‐immunometabolism regulation for strengthening anti-PDL1 therapy against TNBC. Adv Sci (Weinh). 2023;10(3):e2203788. https://doi.org/10.1002/advs.202203788
41. Dong X, Wang X, Zheng X, Jiang H, Liu L, Ma N, et al. Targeted nanoparticle delivery system for tumor-associated macrophage reprogramming to enhance TNBC therapy. Cell Biol Toxicol. 2025;41(1):58. https://doi.org/10.1007/s10565-025-10001-1
42. Yang Y, Li H, Yang W, Shi Y. Improving efficacy of TNBC immunotherapy: based on analysis and subtyping of immune microenvironment. Front Immunol. 2024;15:1441667. https://doi.org/10.3389/fimmu.2024.1441667
43. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16(1):223-249. https://doi.org/10.1146/annurev-pathol-042020-042741
44. Nagasaki J, Ishino T, Togashi Y. Mechanisms of resistance to immune checkpoint inhibitors. Cancer Sci. 2022;113(10):3303-3312. https://doi.org/10.1111/cas.15497
45. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9-16. https://doi.org/10.1038/bjc.2017.434
46. Keenan TE, Tolaney SM. Role of immunotherapy in triple-negative breast cancer. J Natl Compr Canc Netw. 2020;18(4):479-489. https://doi.org/10.6004/jnccn.2020.7554
47. Mediratta K, El-Sahli S, D'Costa V, Wang L. Current progresses and challenges of immunotherapy in triple-negative breast cancer. Cancers (Basel). 2020;12(12):3529. https://doi.org/10.3390/cancers12123529
48. Shen G, Liu Z, Wang M, Zhao Y, Liu X, Hou Y, et al. Neoadjuvant apatinib addition to sintilimab and carboplatin-taxane based chemotherapy in patients with early triple-negative breast cancer: the phase 2 NeoSAC trial. Signal Transduct Target Ther. 2025;10(1):41. https://doi.org/10.1038/s41392-025-02137-7
49. Panagi M, Voutouri C, Mpekris F, Papageorgis P, Martin MR, Martin JD, et al. TGF-β inhibition combined with cytotoxic nanomedicine normalizes triple negative breast cancer microenvironment towards anti-tumor immunity. Theranostics. 2020;10(4):1910-1922. https://doi.org/10.7150/thno.36936
50. Yang M, Guo J, Li J, Wang S, Sun Y, Liu Y, et al. Platycodon grandiflorum-derived extracellular vesicles suppress triple-negative breast cancer growth by reversing the immunosuppressive tumor microenvironment and modulating the gut microbiota. J Nanobiotechnology. 2025;23(1):92. https://doi.org/10.1186/s12951-025-03139-x
51. Wang Q, Li D, Ma H, Li Z, Wu J, Qiao J, et al. Tumor cell-derived EMP1 is essential for cancer-associated fibroblast infiltration in tumor microenvironment of triple-negative breast cancer. Cell Death Dis. 2025;16(1):143. https://doi.org/10.1038/s41419-025-07464-9
52. Bareche Y, Buisseret L, Gruosso T, Girard E, Venet D, Dupont F, et al. Unraveling triple-negative breast cancer tumor microenvironment heterogeneity: towards an optimized treatment approach. J Natl Cancer Inst. 2020;112(7):708-719. https://doi.org/10.1093/jnci/djz208
53. Jiang K, Dong M, Li C, Sheng J. Unraveling heterogeneity of tumor cells and microenvironment and its clinical implications for triple negative breast cancer. Front Oncol. 2021;11:557477. https://doi.org/10.3389/fonc.2021.557477
54. Hu L, Hu J, Qin C, Liu S, Yu Y. Ferroptosis in TNBC: interplay with tumor-infiltrating immune cells and therapeutic implications. Mol Cell Biochem. 2025;480 (9):5029-5039. https://doi.org/10.1007/s11010-025-05305-z
55. Wu C, Dong S, Huang R, Chen X. Cancer-associated adipocytes and breast cancer: intertwining in the tumor microenvironment and challenges for cancer therapy. Cancers (Basel). 2023;15(3):726. https://doi.org/10.3390/cancers15030726
56. Mi H, Varadhan R, Cimino-Mathews AM, Emens LA, Santa-Maria CA, Popel AS. Spatial architecture of single-cell and vasculature in tumor microenvironment predicts clinical outcomes in triple-negative breast cancer. Mod Pathol. 2025;38(2):100652. https://doi.org/10.1016/j.modpat.2024.100652
57. Zhang L, Wang L, Xu Z, Zhang X, Guan S, Liu Z, et al. eNAMPT/Ac-STAT3/DIRAS2 axis promotes development and cancer stemness in triple-negative breast cancer by enhancing cytokine crosstalk between tumor-associated macrophages and cancer cells. Int J Biol Sci. 2025;21(5):2027-2047. https://doi.org/10.7150/ijbs.103723
58. Shu Y, Cheng P. Targeting tumor-associated macrophages for cancer immunotherapy. Biochim Biophys Acta Rev Cancer. 2020;1874(2):188434. https://doi.org/10.1016/j.bbcan.2020.188434
59. Strizova Z, Benesova I, Bartolini R, Novysedlak R, Cecrdlova E, Foley LK, et al. M1/M2 macrophages and their overlaps - myth or reality? Clinical science. 2023;137(15):1067-93. https://doi.org/10.1042/cs20220531
60. Chen Z, Wu J, Wang L, Zhao H, He J. Tumor-associated macrophages of the M1/M2 phenotype are involved in the regulation of malignant biological behavior of breast cancer cells through the EMT pathway. Med Oncol. 2022;39(5):83. https://doi.org/10.1007/s12032-022-01670-7
61. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21(11):799-820. https://doi.org/10.1038/s41573-022-00520-5
62. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787-795. https://doi.org/10.1172/jci59643
63. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv Drug Deliv Rev. 2017;114:206-221. https://doi.org/10.1016/j.addr.2017.04.010
64. Hu Q, Bian Q, Rong D, Wang L, Song J, Huang HS, et al. JAK/STAT pathway: extracellular signals, diseases, immunity, and therapeutic regimens. Front Bioeng Biotechnol. 2023;11:1110765. https://doi.org/10.3389/fbioe.2023.1110765
65. Yuan Y, Wu D, Hou Y, Zhang Y, Tan C, Nie X, et al. Wnt signaling: modulating tumor-associated macrophages and related immunotherapeutic insights. Biochem Pharmacol. 2024;223:116154. https://doi.org/10.1016/j.bcp.2024.116154
66. Bai X, Guo YR, Zhao ZM, Li XY, Dai DQ, Zhang JK, et al. Macrophage polarization in cancer and beyond: from inflammatory signaling pathways to potential therapeutic strategies. Cancer Lett. 2025;625:217772. https://doi.org/10.1016/j.canlet.2025.217772
67. Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer. 2020;19(1):145. https://doi.org/10.1186/s12943-020-01258-7
68. Skytthe MK, Graversen JH, Moestrup SK. Targeting of CD163(+) macrophages in inflammatory and malignant diseases. Int J Mol Sci. 2020;21(15):5497. https://doi.org/10.3390/ijms21155497
69. Allison E, Edirimanne S, Matthews J, Fuller SJ. Breast cancer survival outcomes and tumor-associated macrophage markers: a systematic review and meta-analysis. Oncol Ther. 2023;11(1):27-48. https://doi.org/10.1007/s40487-022-00214-3
70. Jamiyan T, Kuroda H, Yamaguchi R, Abe A, Hayashi M. CD68- and CD163-positive tumor-associated macrophages in triple negative cancer of the breast. Virchows Arch. 2020;477(6):767-775. https://doi.org/10.1007/s00428-020-02855-z
71. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399-416. https://doi.org/10.1038/nrclinonc.2016.217
72. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252-264. https://doi.org/10.1038/nrc3239
73. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. 2017;8:93. https://doi.org/10.3389/fimmu.2017.00093
74. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20(1):131. https://doi.org/10.1186/s12943-021-01428-1
75. Magesh P, Thankachan S, Venkatesh T, Suresh PS. Breast cancer fibroblasts and cross-talk. Clin Chim Acta. 2021;521:158-169. https://doi.org/10.1016/j.cca.2021.07.011
76. Zhao Y, Shen M, Wu L, Yang H, Yao Y, Yang Q, et al. Stromal cells in the tumor microenvironment: accomplices of tumor progression? Cell Death Dis. 2023;14(9):587. https://doi.org/10.1038/s41419-023-06110-6
77. Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47(4):320-329. https://doi.org/10.1038/ng.3225
78. Grout JA, Sirven P, Leader AM, Maskey S, Hector E, Puisieux I, et al. Spatial positioning and matrix programs of cancer-associated fibroblasts promote T-cell exclusion in human lung tumors. Cancer Discov. 2022;12(11):2606-2625. https://doi.org/10.1158/2159-8290.Cd-21-1714
79. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174-186. https://doi.org/10.1038/s41568-019-0238-1
80. Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP, et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis. 2017;6(7):e352. https://doi.org/10.1038/oncsis.2017.54
81. Northcott JM, Dean IS, Mouw JK, Weaver VM. Feeling stress: the mechanics of cancer progression and aggression. Front Cell Dev Biol. 2018;6:17. https://doi.org/10.3389/fcell.2018.00017
82. Wu C, Sun X, Lu Y, Wang H, Yu Z, Wang Z, et al. Cancer-associated fibroblast promotes tamoxifen resistance in estrogen receptor positive breast cancer via exosomal LncRNA PRKCQ-AS1/miR-200a-3p/MKP1 axis-mediated apoptosis suppression. J Exp Clin Cancer Res. 2025;44(1):274. https://doi.org/10.1186/s13046-025-03529-x
83. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15(2):73-79. https://doi.org/10.1038/nrc3876
84. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212-20217. https://doi.org/10.1073/pnas.1320318110
85. Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, et al. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. 2018;11(1):100. https://doi.org/10.1186/s13045-018-0644-y
86. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014;63(7):721-735. https://doi.org/10.1007/s00262-014-1549-4
87. Loi S, Michiels S, Adams S, Loibl S, Budczies J, Denkert C, et al. The journey of tumor-infiltrating lymphocytes as a biomarker in breast cancer: clinical utility in an era of checkpoint inhibition. Ann Oncol. 2021;32(10):1236-1244. https://doi.org/10.1016/j.annonc.2021.07.007
88. Yamashita N, Long M, Fushimi A, Yamamoto M, Hata T, Hagiwara M, et al. MUC1-C integrates activation of the IFN-γ pathway with suppression of the tumor immune microenvironment in triple-negative breast cancer. J Immunother Cancer. 2021;9(1):e002115. https://doi.org/10.1136/jitc-2020-002115
89. Cao Y, Ge X, Zhu X, Han Y, Wang P, Akakuru OU, et al. Transformable neuropeptide prodrug with tumor microenvironment responsiveness for tumor growth and metastasis inhibition of triple‐negative breast cancer. Adv Sci (Weinh). 2023;10(21):2300545. https://doi.org/10.1002/advs.202300545
90. Wang S, Wang Z, Li Z, Xu J, Meng X, Zhao Z, et al. A catalytic immune activator based on magnetic nanoparticles to reprogram the immunoecology of breast cancer from "cold" to "hot" state. Adv Healthc Mater. 2022;11(21):e2201240. https://doi.org/10.1002/adhm.202201240
91. Zhu D, Lu Y, Yan Z, Deng Q, Hu B, Wang Y, et al. A β-carboline derivate PAD4 inhibitor reshapes neutrophil phenotype and improves the tumor immune microenvironment against triple-negative breast cancer. J Med Chem. 2024;67(10):7973-7994. https://doi.org/10.1021/acs.jmedchem.4c00030
92. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21(8):485-498. https://doi.org/10.1038/s41577-020-00490-y
93. Huang P, Zhou X, Zheng M, Yu Y, Jin G, Zhang S. Regulatory T cells are associated with the tumor immune microenvironment and immunotherapy response in triple-negative breast cancer. Front Immunol. 2023;14:1263537. https://doi.org/10.3389/fimmu.2023.1263537
94. Kang JH, Zappasodi R. Modulating Treg stability to improve cancer immunotherapy. Trends Cancer. 2023;9(11):911-927. https://doi.org/10.1016/j.trecan.2023.07.015
95. Malla RR, Vasudevaraju P, Vempati RK, Rakshmitha M, Merchant N, Nagaraju GP. Regulatory T cells: their role in triple-negative breast cancer progression and metastasis. Cancer. 2022;128(6):1171-1183. https://doi.org/10.1002/cncr.34084
96. Zheng C, Xu X, Wu M, Xue L, Zhu J, Xia H, et al. Neutrophils in triple-negative breast cancer: an underestimated player with increasingly recognized importance. Breast Cancer Res. 2023;25(1):88. https://doi.org/10.1186/s13058-023-01676-7
97. SenGupta S, Hein LE, Xu Y, Zhang J, Konwerski JR, Li Y, et al. Triple-negative breast cancer cells recruit neutrophils by secreting TGF-β and CXCR2 ligands. Front Immunol. 2021;12:659996. https://doi.org/10.3389/fimmu.2021.659996
98. Munkácsy G, Santarpia L, Győrffy B. Therapeutic potential of tumor metabolic reprogramming in triple-negative breast cancer. Int J Mol Sci. 2023;24(8):6945. https://doi.org/10.3390/ijms24086945
99. Abdel-Latif M, Youness RA. Why natural killer cells in triple negative breast cancer? World J Clin Oncol. 2020;11(7):464-476. https://doi.org/10.5306/wjco.v11.i7.464
100. Zhang Y, Ji S, Miao G, Du S, Wang H, Yang X, et al. The current role of dendritic cells in the progression and treatment of colorectal cancer. Cancer Biol Med. 2024;21(9):769-783. https://doi.org/10.20892/j.issn.2095-3941.2024.0188
101. Hu X, Dou Q, Jiang P, Zhang M, Wang J. Targeting matrix metalloproteinases activating and indoleamine 2,3-dioxygenase suppression for triple-negative breast cancer multimodal therapy. Int J Biol Macromol. 2025;310(Part 3):143289. https://doi.org/10.1016/j.ijbiomac.2025.143289
102. Lu X, Gou Z, Chen H, Li L, Chen F, Bao C, et al. Extracellular matrix cancer-associated fibroblasts promote stromal fibrosis and immune exclusion in triple-negative breast cancer. J Pathol. 2025;265(3):385-399. https://doi.org/10.1002/path.6395
103. Zhang Y, Zhou J, Wang Y, Wu Y, Li Y, Wang B, et al. Stimuli-responsive polymer-dasatinib prodrug to reprogram cancer-associated fibroblasts for boosted immunotherapy. J Control Release. 2025;381:113606. https://doi.org/10.1016/j.jconrel.2025.113606
104. Vecchi L, Mota STS, Zóia MAP, Martins IC, Souza JB, Santos TG, et al. Interleukin-6 signaling in triple negative breast cancer cells elicits the annexin A1/formyl peptide receptor 1 axis and affects the tumor microenvironment. Cells. 2022;11(10):1705. https://doi.org/10.3390/cells11101705
105. Yang M, Qin C, Tao L, Cheng G, Li J, Lv F, et al. Synchronous targeted delivery of TGF-β siRNA to stromal and tumor cells elicits robust antitumor immunity against triple-negative breast cancer by comprehensively remodeling the tumor microenvironment. Biomaterials. 2023;301:122253. https://doi.org/10.1016/j.biomaterials.2023.122253
106. Tsai TH, Yang CC, Kou TC, Yang CE, Dai JZ, Chen CL, et al. Overexpression of GLUT3 promotes metastasis of triple‐negative breast cancer by modulating the inflammatory tumor microenvironment. J Cell Physiol. 2021;236(6):4669-4680. https://doi.org/10.1002/jcp.30189
107. Ren X, Cheng Z, He J, Yao X, Liu Y, Cai K, et al. Inhibition of glycolysis-driven immunosuppression with a nano-assembly enhances response to immune checkpoint blockade therapy in triple negative breast cancer. Nat Commun. 2023;14(1):7021. https://doi.org/10.1038/s41467-023-42883-2
108. Xie Y, Xie F, Zhou X, Zhang L, Yang B, Huang J, et al. Microbiota in tumors: from understanding to application. Adv Sci (Weinh). 2022;9(21):e2200470. https://doi.org/10.1002/advs.202200470
109. Fu A, Yao B, Dong T, Chen Y, Yao J, Liu Y, et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell. 2022;185(8):1356-72.e26. https://doi.org/10.1016/j.cell.2022.02.027
110. Devoy C, Flores Bueso Y, Tangney M. Understanding and harnessing triple-negative breast cancer-related microbiota in oncology. Front Oncol. 2022;12:1020121. https://doi.org/10.3389/fonc.2022.1020121
111. Liao K, Wen J, Liu Z, Zhang B, Zhang X, Fu Y, et al. The role of intratumoral microbiome in the occurrence, proliferation, metastasis of colorectal cancer and its underlying therapeutic strategies. Ageing Res Rev. 2025;111:102820. https://doi.org/10.1016/j.arr.2025.102820
112. Parhi L, Alon-Maimon T, Sol A, Nejman D, Shhadeh A, Fainsod-Levi T, et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun. 2020;11(1):3259. https://doi.org/10.1038/s41467-020-16967-2
113. Akbari E, Epstein JB, Samim F. Unveiling the hidden links: periodontal disease, fusobacterium nucleatum, and cancers. Curr Oncol Rep. 2024;26(11):1388-1397. https://doi.org/10.1007/s11912-024-01591-w
114. Parida S, Wu S, Siddharth S, Wang G, Muniraj N, Nagalingam A, et al. A procarcinogenic colon microbe promotes breast tumorigenesis and metastatic progression and concomitantly activates notch and β-catenin axes. Cancer Discov. 2021;11(5):1138-1157. https://doi.org/10.1158/2159-8290.Cd-20-0537
115. Chiba A, Bawaneh A, Velazquez C, Clear KYJ, Wilson AS, Howard-McNatt M, et al. Neoadjuvant chemotherapy shifts breast tumor microbiota populations to regulate drug responsiveness and the development of metastasis. Mol Cancer Res. 2020;18(1):130-139. https://doi.org/10.1158/1541-7786.Mcr-19-0451
116. Li T, Huang Y, Cui S, Hong Z, Zhang X, Li Z, et al. RNA methylation patterns of tumor microenvironment cells regulate prognosis and immunotherapeutic responsiveness in patients with triple-negative breast cancer. Sci Rep. 2024;14(1):26075. https://doi.org/10.1038/s41598-024-77941-2
117. Adewunmi O, Shen Y, Zhang XHF, Rosen JM. Targeted inhibition of lncRNA malat1 alters the tumor immune microenvironment in preclinical syngeneic mouse models of triple-negative breast cancer. Cancer Immunol Res. 2023;11(11):1462-1479. https://doi.org/10.1158/2326-6066.cir-23-0045
118. Sabit H, Adel A, Abdelfattah MM, Ramadan RM, Nazih M, Abdel-Ghany S, et al. The role of tumor microenvironment and immune cell crosstalk in triple-negative breast cancer (TNBC): emerging therapeutic opportunities. Cancer Lett. 2025;628:217865. https://doi.org/10.1016/j.canlet.2025.217865
119. Tahtaci G, Gunel N, Sadioglu A, Akyurek N, Boz O, Uner A. LAG-3 expression in tumor microenvironment of triple-negative breast cancer. Turk J Med Sci. 2023;53(1):142–148. https://doi.org/10.55730/1300-0144.5567
120. Xiong W, Cheng Z, Chen H, Liang H, Wang M, Chen Y, et al. Biomimetic tumor cell membrane‐encapsulated nanoparticles combine NIR‐II photothermal therapy and chemotherapy for enhanced immunotherapy in triple-negative breast cancer. Adv Funct Mater. 2024;34(52):2410841. https://doi.org/10.1002/adfm.202410841
121. Farshbafnadi M, Pastaki Khoshbin A, Rezaei N. Immune checkpoint inhibitors for triple-negative breast cancer: from immunological mechanisms to clinical evidence. Int Immunopharmacol. 2021;98:107876. https://doi.org/10.1016/j.intimp.2021.107876
122. Nguyen HM, Paulishak W, Oladejo M, Wood L. Dynamic tumor microenvironment, molecular heterogeneity, and distinct immunologic portrait of triple-negative breast cancer: an impact on classification and treatment approaches. Breast Cancer. 2023;30(2):167-186. https://doi.org/10.1007/s12282-022-01415-4
123. Debien V, De Caluwé A, Wang X, Piccart-Gebhart M, Tuohy VK, Romano E, et al. Immunotherapy in breast cancer: an overview of current strategies and perspectives. NPJ Breast Cancer. 2023;9(1):7. https://doi.org/10.1038/s41523-023-00508-3
124. Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(1):44-59. https://doi.org/10.1016/s1470-2045(19)30689-8
125. Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol. 2022;23(8):1148-1156. https://doi.org/10.1038/s41590-022-01267-2
126. Jiang R, Yang L, Liu X, Xu Y, Han L, Chen Y, et al. Genetically engineered macrophages reverse the immunosuppressive tumor microenvironment and improve immunotherapeutic efficacy in TNBC. Mol Ther. 2025;33(7):3339-3359. https://doi.org/10.1016/j.ymthe.2025.03.024
127. Kalinski P, Kokolus KM, Gandhi S. Paclitaxel, interferons and functional reprogramming of tumor-associated macrophages in optimized chemo-immunotherapy. J Immunother Cancer. 2025;13(5):e010960. https://doi.org/10.1136/jitc-2024-010960
128. He L, Jhong JH, Chen Q, Huang KY, Strittmatter K, Kreuzer J, et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. 2021;37(5):109955. https://doi.org/10.1016/j.celrep.2021.109955
129. Fan J, Qin Y, Qiu W, Liang J, Xiao C, Xie Q, et al. Gamabufotalin loaded micro-nanocomposites for multimodal therapy of metastatic TNBC by efficiently inducing ICD. Biomaterials. 2025;314:122851. https://doi.org/10.1016/j.biomaterials.2024.122851
130. Chen M, Song L, Zhou Y, Xu T, Sun T, Liu Z, et al. Promotion of triple negative breast cancer immunotherapy by combining bioactive radicals with immune checkpoint blockade. Acta Biomater. 2025;194:305-322. https://doi.org/10.1016/j.actbio.2025.01.015
131. Chen X, Yang M, Yin J, Li P, Zeng S, Zheng G, et al. Tumor-associated macrophages promote epithelial-mesenchymal transition and the cancer stem cell properties in triple-negative breast cancer through CCL2/AKT/β-catenin signaling. Cell Commun Signal. 2022;20(1):92. https://doi.org/10.1186/s12964-022-00888-2
132. Colligan SH, Amitrano AM, Zollo RA, Peresie J, Kramer ED, Morreale B, et al. Inhibiting the biogenesis of myeloid-derived suppressor cells enhances immunotherapy efficacy against mammary tumor progression. J Clin Invest. 2022;132(23):e158661. https://doi.org/10.1172/jci158661
133. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221:107753. https://doi.org/10.1016/j.pharmthera.2020.107753
134. Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan C-L, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9(1):2897. https://doi.org/10.1038/s41467-018-05220-6
135. Lim SA. Metabolic reprogramming of the tumor microenvironment to enhance immunotherapy. BMB Rep. 2024;57(9):388-399. https://doi.org/10.5483/BMBRep.2024-0031
136. Yang X, Cui X, Wang G, Zhou M, Wu Y, Du Y, et al. HDAC inhibitor regulates the tumor immune microenvironment via pyroptosis in triple negative breast cancer. Mol Carcinog. 2024;63(9):1800-1813. https://doi.org/10.1002/mc.23773
137. Liu X, Sun M, Pu F, Ren J, Qu X. Transforming intratumor bacteria into immunopotentiators to reverse cold tumors for enhanced immuno-chemodynamic therapy of triple-negative breast cancer. J Am Chem Soc. 2023;145(48):26296-26307. https://doi.org/10.1021/jacs.3c09472
138. Kim S, Han I-H, Lee S, Park D, Lee H, Kim J, et al. The combination of CD300c antibody with PD-1 blockade suppresses tumor growth and metastasis by remodeling the tumor microenvironment in triple-negative breast cancer. Int J Mol. Sci. 2025;26(11):5045. https://doi.org/10.3390/ijms26115045
139. Liu L, He H, Luo Z, Zhou H, Liang R, Pan H, et al. In situ photocatalyzed oxygen generation with photosynthetic bacteria to enable robust immunogenic photodynamic therapy in triple‐negative breast cancer. Adv Funct Mater. 2020;30(10):1910176. https://doi.org/10.1002/adfm.201910176
140. Fang K, Yuan S, Zhang X, Zhang J, Sun SL, Li X. Regulation of immunogenic cell death and potential applications in cancer therapy. Front Immunol. 2025;16:1571212. https://doi.org/10.3389/fimmu.2025.1571212
141. Loibl S, Schneeweiss A, Huober J, Braun M, Rey J, Blohmer JU, et al. Neoadjuvant durvalumab improves survival in early triple-negative breast cancer independent of pathological complete response. Ann Oncol. 2022;33(11):1149-1158. https://doi.org/10.1016/j.annonc.2022.07.1940
142. Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396(10265):1817-1828. https://doi.org/10.1016/s0140-6736(20)32531-9
143. Tezcan O, Elshafei AS, Benderski K, Rama E, Wagner M, Moeckel D, et al. Effect of cellular and microenvironmental multidrug resistance on tumor-targeted drug delivery in triple-negative breast cancer. J Control Release. 2023;354:784-793. https://doi.org/10.1016/j.jconrel.2022.12.056
144. Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res. 2019;25(18):5449-5457. https://doi.org/10.1158/1078-0432.Ccr-18-1543
145. Liu J, Liu Q, Li Y, Li Q, Su F, Yao H, et al. Efficacy and safety of camrelizumab combined with apatinib in advanced triple-negative breast cancer: an open-label phase II trial. J Immunother Cancer. 2020;8(1):e000696. https://doi.org/10.1136/jitc-2020-000696
146. Vito A, Rathmann S, Mercanti N, El-Sayes N, Mossman K, Valliant J. Combined radionuclide therapy and immunotherapy for treatment of triple negative breast cancer. Int J Mol Sci. 2021;22(9):4843. https://doi.org/10.3390/ijms22094843
147. Chen X, Feng L, Huang Y, Wu Y, Xie N. Mechanisms and strategies to overcome PD-1/PD-L1 blockade resistance in triple-negative breast cancer. Cancers (Basel). 2022;15(1):104. https://doi.org/10.3390/cancers15010104
148. Castellano G, Giugliano F, Curigliano G, Marra A. Clinical utility of genomic signatures for the management of early and metastatic triple-negative breast cancer. Curr Opin Oncol. 2023;35(6):479-490. https://doi.org/10.1097/cco.0000000000000989
149. Golden EB, Marciscano AE, Formenti SC. Radiation therapy and the in situ vaccination approach. Int J Radiat Oncol Biol Phys. 2020;108(4):891-898. https://doi.org/10.1016/j.ijrobp.2020.08.023
150. Cao Y, Meng F, Cai T, Gao L, Lee J, Solomevich SO, et al. Nanoparticle drug delivery systems responsive to tumor microenvironment: promising alternatives in the treatment of triple‐negative breast cancer. WIREs Nanomed Nanobiotechnol. 2024;16(2):e1950. https://doi.org/10.1002/wnan.1950
151. Zhen X, Li Y, Yuan W, Zhang T, Li M, Huang J, et al. Biointerface‐engineered hybrid nanovesicles for targeted reprogramming of tumor microenvironment. Adv Mater. 2024;36(41):2401495. https://doi.org/10.1002/adma.202401495
152. Yu L, Gao L, Liang B, Zhang L, Wu M, Liu J. Polymer-based nanodrugs enhance sonodynamic therapy through epigenetic reprogramming of the immunosuppressive tumor microenvironment. J Control Release. 2025;380:125-137. https://doi.org/10.1016/j.jconrel.2025.01.086
153. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711-3720. https://doi.org/10.1158/1078-0432.Ccr-16-3215
154. Wang S, Sun K, Xiao Y, Feng B, Mikule K, Ramaswamy S, et al. Evaluation of niraparib in combination with anti-PD-1/anti-PD-L1 in preclinical models. Cancer Res. 2018;78(13): Abstract No. 1724. https://doi.org/10.1158/1538-7445.am2018-1724
155. Wang S, Chang CW, Huang J, Zeng S, Zhang X, Hung MC, et al. Gasdermin C sensitizes tumor cells to PARP inhibitor therapy in cancer models. J Clin Invest. 2024;134(1):e166841. https://doi.org/10.1172/jci166841
156. Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, et al. Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA Oncol. 2019;5(8):1132-1140. https://doi.org/10.1001/jamaoncol.2019.1029
157. Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21(9):1155-1164. https://doi.org/10.1016/s1470-2045(20)30324-7
158. Rugo H, Robson M, Im SA, Dalenc F, Ruiz EY, Im YH, et al. Pembrolizumab + olaparib vs pembrolizumab + chemotherapy after induction with pembrolizumab + chemotherapy for locally recurrent inoperable or metastatic TNBC: randomized open-label phase 2 KEYLYNK-009 study. Cancer Res. 2024;84(9):GS01-5-GS-5. https://doi.org/10.1158/1538-7445.SABCS23-GS01-05
159. Vonderheide RH, Domchek SM, Clark AS. Immunotherapy for Breast Cancer: What Are We Missing? Clin Cancer Res. 2017;23(11):2640-6. https://doi.org/10.1158/1078-0432.Ccr-16-2569
160. Zachariah NN, Basu A, Gautam N, Ramamoorthi G, Kodumudi KN, Kumar NB, et al. Intercepting premalignant, preinvasive breast lesions through vaccination. Front Immunol. 2021;12:786286. https://doi.org/10.3389/fimmu.2021.786286
161. Abdou Y, Goudarzi A, Yu JX, Upadhaya S, Vincent B, Carey LA. Immunotherapy in triple negative breast cancer: beyond checkpoint inhibitors. NPJ Breast Cancer. 2022;8(1):121. https://doi.org/10.1038/s41523-022-00486-y
162. Rosenbaum SR, Hughes CJ, Fields KM, Purdy SC, Gustafson AL, Wolin A, et al. EYA3 regulation of NF-κb and CCL2 suppresses cytotoxic NK cells in the premetastatic niche to promote TNBC metastasis. Sci Adv. 2025;11(19):eadt0504. https://doi.org/10.1126/sciadv.adt0504
163. Wu S, Ge A, Deng X, Liu L, Wang Y. Evolving immunotherapeutic solutions for triple-negative breast carcinoma. Cancer Treat Rev. 2024;130:102817. https://doi.org/10.1016/j.ctrv.2024.102817
164. Ravi K, Zhang Y, Sakala L, Manoharan TJM, Pockaj B, LaBaer J, et al. Tumor microenvironment on-a-chip and single-cell analysis reveal synergistic stromal-immune crosstalk on breast cancer progression. Adv Sci (Weinh). 2025;12(16):e2413457. https://doi.org/10.1002/advs.202413457
165. González-Callejo P, García-Astrain C, Herrero-Ruiz A, Henriksen-Lacey M, Seras-Franzoso J, Abasolo I, et al. 3D bioprinted tumor-stroma models of triple-negative breast cancer stem cells for preclinical targeted therapy evaluation. ACS Appl Mater Interfaces. 2024;16(21):27151-27163. https://doi.org/10.1021/acsami.4c04135
166. Doherty MR, Parvani JG, Tamagno I, Junk DJ, Bryson BL, Cheon HJ, et al. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019;21(1):54. https://doi.org/10.1186/s13058-019-1136-x
167. Hua Z, White J, Zhou J. Cancer stem cells in TNBC. Semin Cancer Biol. 2022;82:26-34. https://doi.org/10.1016/j.semcancer.2021.06.015
168. Jan A, Sofi S, Jan N, Mir MA. An update on cancer stem cell survival pathways involved in chemoresistance in triple-negative breast cancer. Future Oncol. 2025;21(6):715-735. https://doi.org/10.1080/14796694.2025.2461443
169. Liu CC, Chen L, Cai YW, Chen YF, Liu YM, Zhou YJ, et al. Targeting EMSY-mediated methionine metabolism is a potential therapeutic strategy for triple-negative breast cancer. Cell Rep Med. 2024;5(2):101396. https://doi.org/10.1016/j.xcrm.2024.101396
170. Liu M, Qian M, Sun W, Sun X, Sun Y, Yu M, et al. Immunosuppressive microenvironment of liver restrains chemotherapeutic efficacy in triple-negative breast cancer. J Immunother Cancer. 2025;13(3):e010871. https://doi.org/10.1136/jitc-2024-010871
171. Zheng P, Hu Z, Shen Y, Gu L, Ouyang Y, Duan Y, et al. PSAT1 impairs ferroptosis and reduces immunotherapy efficacy via GPX4 hydroxylation. Nat Chem Biol. 2025;21:1420-1432. https://doi.org/10.1038/s41589-025-01887-3
172. Jing Y, Wu Y, Hu Q, Wu W, Li D, Hu W, et al. Remodelling hypoxic TNBC microenvironment restores antitumor efficacy of Vγ9Vδ2 T cell therapy. Br J Cancer. 2025;133(3):365-380. https://doi.org/10.1038/s41416-025-03045-x
173. Wang M, Zheng Y, Hao Q, Mao G, Dai Z, Zhai Z, et al. Hypoxic BMSC-derived exosomal miR-210-3p promotes progression of triple-negative breast cancer cells via NFIX-Wnt/β-catenin signaling axis. J Transl Med. 2025;23(1):39. https://doi.org/10.1186/s12967-024-05947-5
174. El-Guindy DM, Ibrahim FM, Ali DA, El-Horany HE, Sabry NM, Elkholy RA, et al. Hypoxia-induced autophagy in triple negative breast cancer: association with prognostic variables, patients' survival and response to neoadjuvant chemotherapy. Virchows Arch. 2023;482(5):823-837. https://doi.org/10.1007/s00428-023-03527-4
175. Zhu Y, Zhu X, Tang C, Guan X, Zhang W. Progress and challenges of immunotherapy in triple-negative breast cancer. Biochim Biophys Acta Rev Cancer. 2021;1876(2):188593. https://doi.org/10.1016/j.bbcan.2021.188593
176. Tiwari H, Singh S, Sharma S, Gupta P, Verma A, Chattopadhaya A, et al. Deciphering the landscape of triple negative breast cancer from microenvironment dynamics and molecular insights to biomarker analysis and therapeutic modalities. Med Res Rev. 2025;45(3):817-841. https://doi.org/10.1002/med.22090
177. Carlino F, Diana A, Piccolo A, Ventriglia A, Bruno V, De Santo I, et al. Immune-based therapy in triple-negative breast cancer: from molecular biology to clinical practice. Cancers (Basel). 2022;14(9):2102. https://doi.org/10.3390/cancers14092102
178. Lindberg I, Saleh A, Tutzauer J, Gunnarsdottir FB, Rydén L, Bergenfelz C, et al. Prognostic relevance of CD163+ immune cells in patients with metastatic breast cancer. Cancer Immunol Immunother. 2025;74(2):42. https://doi.org/10.1007/s00262-024-03892-2
179. Chen Y, Yang L, Huang Y, Zhu T, Zhang L, Cheng M, et al. Intratumoral microbiota predicts the response to neoadjuvant chemoimmunotherapy in triple-negative breast cancer. J Immunother Cancer. 2025;13(4):e010365. https://doi.org/10.1136/jitc-2024-010365
180. Marcel V, Ghayad SE, Belin S, Therizols G, Morel AP, Solano-Gonzàlez E, et al. P53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell. 2013;24(3):318–330. https://doi.org/10.1016/j.ccr.2013.08.013
181. Tang Y, Xu A, Xu Z, Xie J, Huang W, Zhang L, et al. Multi-omics analyses of the heterogenous immune microenvironment in triple-negative breast cancer implicate UQCRFS1 potentiates tumor progression. Exp Hematol Oncol. 2025;14(1):85. https://doi.org/10.1186/s40164-025-00672-1
182. Mariano NC, Marotti JD, Chen Y, Karakyriakou B, Salgado R, Christensen BC, et al. Quantitative proteomics analysis of triple-negative breast cancers. NPJ Precis Oncol. 2025;9(1):117. https://doi.org/10.1038/s41698-025-00907-8
Declarations
Funding Statement: This work was supported by the National Natural Science Foundation of China (No. 81502260), the Organization Department of Shanghai Municipal Party Committee, Shanghai Science and Technology Commission (No. 23Y11909200 and No. 24SF1900200), Shanghai Xuhui District Science and Technology Committee (No. 23XHYD-06), Chinese Society of Clinical Oncology Foundation (No. Y-2022HER-2AZMS-0349 and No. Y-2023AZMETMS-0077), and Xiamen Haicang District Bureau of Science, Technology, Industry, Information Technology and Commerce (No. 350205Z20242010). We thank the National Natural Science Foundation of China, the Organization Department of the Shanghai Municipal Party Committee, the Shanghai Xuhui District Science and Technology Committee, the Chinese Society of Clinical Oncology Foundation, and the Xiamen Haicang District Bureau of Science, Technology, Industry, Information Technology, and Commerce.
Declaration of Competing Interest: The authors declare no financial or personal relationships with other individuals or organizations that could inappropriately influence or bias the content of this work. All authors have read the final version of the manuscript and confirm that there are no competing interests.
Consent for publication: All authors have approved the final version of the manuscript.
Use of Artificial Intelligence (AI) Disclosure: This article was written by human contributors. Artificial intelligence-based tools were used to improve grammar, language, and readability without affecting the article's scientific content, data interpretation, or conclusions. The authors reviewed and verified all content to ensure its accuracy and integrity.
Dual Use Research of Concern (DURC): “The authors confirm that this research does not constitute DURC as defined by the U.S. Government Policy for Oversight of Life Sciences DURC.”
Data Availability Statements (DAS): “No datasets were generated or analyzed in the current study.”
Ethics approval and consent to participate: Not applicable, as this study did not involve the conduct of research.
Authors’ affiliations
1. Department of Medical Oncology, Fudan University Shanghai Cancer Center, Xiamen Hospital, Xiamen, China
2. Phase I Unit, Fudan University Shanghai Cancer Center, Shanghai, China
3. Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China
4. Department of Oncology, The Second People's Hospital of Kashi, Kashi, Xinjiang, China
# Correspondence: Wenxing Qin - Email: qinwenxingqwx@163.com
* Youli Li, Yang Chen, Yuxin Mu, and Xuemei Xiu are equal contributors to this work and are designated as co-first authors.
CRediT authorship contribution statement: Youli Li: Writing – original draft, Writing – review & editing. Yang Chen: Writing – original draft, Writing – review & editing. Yuxin Mu: Writing – review & editing. Xuemei Xiu: Writing – original draft, Writing – review & editing. Wenxind Qin: Conceptualization, Resources, Project administration, Supervision, Writing – original draft, Writing – review & editing. All authors contributed to the work and approved the final version of the manuscript.
ORCID ID
Youli Li: https://orcid.org/0000-0003-3644-3930
Yang Chen: https://orcid.org/0000-0003-2162-3648
Yuxin Mu: https://orcid.org/0000-0002-8465-6933
Xuemei Xiu: https://orcid.org/0009-0007-1031-0503
Wenxing Qin: https://orcid.org/0009-0002-7274-4917
How to cite
Li Y, Chen Y, Mu Y, Xiu X, Qin W. Tumor Microenvironment in Triple-Negative Breast Cancer and Targeting Approaches. Cancer Biome and Targeted Therapy. Published online 2025 Dec.